5月29日�����,國家藥監(jiān)局審評中心發(fā)布了《已上市血液制品生產(chǎn)場地變更研究技術(shù)指導(dǎo)原則(試行)》���,該文件旨在指導(dǎo)血液制品上市許可持有人(以下簡稱持有人)科學(xué)規(guī)范開展血液制品上市后場地變更藥學(xué)研究���,引導(dǎo)和促進(jìn)血液制品持有人利用場地變更進(jìn)行生產(chǎn)工藝升級優(yōu)化和硬件系統(tǒng)改造��,加強(qiáng)對已上市血液制品藥學(xué)變更的管理。文件自發(fā)布之日起施行。
為指導(dǎo)血液制品上市許可持有人科學(xué)規(guī)范開展血液制品上市后場地變更藥學(xué)研究���,引導(dǎo)和促進(jìn)血液制品持有人利用場地變更進(jìn)行生產(chǎn)工藝升級優(yōu)化和硬件系統(tǒng)改造,加強(qiáng)對已上市血液制品藥學(xué)變更的管理�����,藥審中心組織制定了《已上市血液制品生產(chǎn)場地變更研究技術(shù)指導(dǎo)原則(試行)》���。根據(jù)《國家藥監(jiān)局綜合司關(guān)于印發(fā)藥品技術(shù)指導(dǎo)原則發(fā)布程序的通知》(藥監(jiān)綜藥管〔2020〕9號)要求,經(jīng)國家藥品監(jiān)督管理局審查同意�,現(xiàn)予發(fā)布���,自發(fā)布之日起施行�����。
特此通告�����。
已上市血液制品生產(chǎn)場地變更研究技術(shù)指導(dǎo)原則(試行)
目 錄
一����、前言
二、基本考量
1���、科學(xué)規(guī)劃
2����、場地變更的基本要求
(1)血漿來源控制
(2)病毒去除/滅活及病毒安全性風(fēng)險(xiǎn)評估
(3)工藝驗(yàn)證
3、可比性研究
4、溝通交流
三���、變更類型事項(xiàng)及技術(shù)要求
四����、其他考量
1、增加生產(chǎn)場地/生產(chǎn)線
2��、非臨床研究及臨床試驗(yàn)
3、風(fēng)險(xiǎn)管控
五����、參考文獻(xiàn)
一���、前言
為指導(dǎo)血液制品上市許可持有人(以下簡稱持有人)科學(xué)規(guī)范開展血液制品上市后場地變更藥學(xué)研究�����,引導(dǎo)和促進(jìn)血液制品持有人利用場地變更進(jìn)行生產(chǎn)工藝升級優(yōu)化和硬件系統(tǒng)改造,加強(qiáng)對已上市血液制品藥學(xué)變更的管理,按照《中華人民共和國藥品管理法》�����、《藥品注冊管理辦法》�、《藥品生產(chǎn)監(jiān)督管理辦法》、《藥品上市后變更管理辦法(試行)》�����、《中國藥典》�,以及《血液制品管理?xiàng)l例》等相關(guān)規(guī)定和要求�����,制定本指導(dǎo)原則�����。
本指導(dǎo)原則適用于人血漿來源的血液制品���,對于動(dòng)物血源產(chǎn)品(如馬免疫血清制品��、豬源纖維蛋白粘合劑等)��,可參考國內(nèi)外其他相關(guān)指導(dǎo)原則并借鑒本指導(dǎo)原則的基本理念開展相應(yīng)變更研究��。
本指導(dǎo)原則所稱血液制品場地變更是指血液制品上市后�����,持 有人于異地或原址新建�、擴(kuò)建或改建生產(chǎn)廠房���,通常涉及一個(gè)或多個(gè)品種的場地轉(zhuǎn)移�����。本指導(dǎo)原則系在《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》基礎(chǔ)上�,針對血液制品的特殊性�,圍繞血液制品場地變更存在的突出問題起草。本指導(dǎo)原則范圍內(nèi)的變更���,以本指導(dǎo)原則為準(zhǔn)��;本指導(dǎo)原則范圍之外的其他藥學(xué)變更����,可參考《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》。對于另有規(guī)定和技術(shù)要求的�����,也應(yīng)遵照執(zhí)行�����。
嚴(yán)格實(shí)施藥品生產(chǎn)質(zhì)量管理規(guī)范(GMP)和建立有效的藥品質(zhì)量管理體系(PQS)是執(zhí)行血液制品上市后藥學(xué)變更的前提和必要條件��。持有人是血液制品上市后變更管理的責(zé)任主體���,承擔(dān)血液制品全生命周期管理義務(wù),應(yīng)當(dāng)按照藥品監(jiān)管法律法規(guī)的相關(guān)要求��,建立血液制品場地變更控制體系���,對血液制品場地變更及其關(guān)聯(lián)變更藥學(xué)研究的科學(xué)性�、完整性���、真實(shí)性負(fù)責(zé)���。鼓勵(lì)持有人不斷改進(jìn)和優(yōu)化生產(chǎn)工藝�,持續(xù)提高產(chǎn)品質(zhì)量�����,開展充分的驗(yàn)證和可比性研究以證明變更不會(huì)對產(chǎn)品的安全性�、有效性和質(zhì)量可控性產(chǎn)生不良影響。
本指導(dǎo)原則是對血液制品場地變更及其關(guān)聯(lián)變更藥學(xué)研究 的技術(shù)要求�����。持有人應(yīng)基于具體變更事項(xiàng)及研究數(shù)據(jù)自行開展風(fēng)險(xiǎn)評估�,參考相關(guān)技術(shù)指導(dǎo)原則,確定變更分類和申報(bào)途徑�����。在充分研究�、評估和必要的驗(yàn)證基礎(chǔ)上仍無法確定變更管理類別的,鼓勵(lì)持有人按照《藥品上市后變更管理辦法(試行)》通過溝通交流途徑與相應(yīng)藥品監(jiān)管部門及技術(shù)單位達(dá)成一致�����。本指導(dǎo)原則僅反映當(dāng)前對血液制品的科學(xué)認(rèn)知���,隨著對產(chǎn)品 認(rèn)識的不斷深入及政策法規(guī)的變化��,將不斷進(jìn)行相應(yīng)更新和完善�����。
二�、基本考量
1、科學(xué)規(guī)劃
由于不同血液制品自身特點(diǎn)不同���,變更事項(xiàng)、變更程度及先驗(yàn)知識等不同�,變更帶來的潛在風(fēng)險(xiǎn)也會(huì)有差別。為保證變更研究有序開展���,減少變更造成的非預(yù)期影響���,應(yīng)在場地變更前對藥學(xué)研究和變更策略進(jìn)行科學(xué)規(guī)劃和管理。持有人應(yīng)具備足夠的知識積累��,具備風(fēng)險(xiǎn)識別�、風(fēng)險(xiǎn)評估和風(fēng)險(xiǎn)管控能力。在實(shí)施變更時(shí)持有人應(yīng)基于變更和風(fēng)險(xiǎn)控制策略���,充分開展研究和驗(yàn)證�����。既往產(chǎn)品注冊階段��、以及上市后生產(chǎn)過程中積累的研究和生產(chǎn)數(shù)據(jù)是血液制品場地變更研究的基礎(chǔ)����。研究工作越系統(tǒng)���、越深入�����,生產(chǎn)過程中積累的數(shù)據(jù)越充分����,對血液制品場地變更的研究和風(fēng)險(xiǎn)評估越有幫助�。
鼓勵(lì)持有人一次性完成血液制品品種整體場地變更,若因品種較多和/或生產(chǎn)場地���、設(shè)施設(shè)備����、人員受限等特殊情況無法一次性完成所有品種的場地變更�,持有人應(yīng)根據(jù)產(chǎn)品特點(diǎn)及生產(chǎn)情況,科學(xué)規(guī)劃����、合理安排品種場地轉(zhuǎn)移次序及階段�,首個(gè)轉(zhuǎn)移階段是整體變更的基礎(chǔ),需進(jìn)行全面���、系統(tǒng)的變更研究,建議考慮轉(zhuǎn)移產(chǎn)品生產(chǎn)工藝���、規(guī)格、規(guī)模等方面的代表性����,并開展首個(gè)轉(zhuǎn)移階段對后續(xù)轉(zhuǎn)移階段支持性的論證;后續(xù)轉(zhuǎn)移階段品種的驗(yàn)證程度���、研究內(nèi)容可基于首個(gè)階段的研究結(jié)果經(jīng)評估后確定�����。
鼓勵(lì)持有人盡快完成信息化系統(tǒng)的建立���,增強(qiáng)血漿來源、生產(chǎn)及檢驗(yàn)過程中的全面可溯源性及真實(shí)性監(jiān)測��。
通常場地變可能涉及生產(chǎn)設(shè)備變更,持有人應(yīng)充分評估設(shè)備差異對工藝參數(shù)實(shí)際輸出能力和控制范圍的影響����,若因設(shè)備差異等導(dǎo)致工藝參數(shù)輸出能力和控制范圍超出變更前�����,申請人應(yīng)開展充分的研究�����,以評估影響����,確保變更后工藝控制能力不降低。
2��、場地變更的基本要求
(1) 血漿來源控制
進(jìn)行血液制品生產(chǎn)場地變更時(shí)��,若涉及新增或變更漿站�����,
或涉及漿站設(shè)置主體變更的���,均應(yīng)符合相關(guān)要求。
鼓勵(lì)持有人建立原料血漿集中質(zhì)量體系管理下的檢驗(yàn)中心���,采用先進(jìn)檢測技術(shù)加強(qiáng)對不同漿站、不同生產(chǎn)地點(diǎn)原料血漿檢驗(yàn)檢測體系的統(tǒng)一管控��,確保檢測體系的規(guī)范和完善����。
(2) 病毒去除/滅活及病毒安全性風(fēng)險(xiǎn)評估
【血漿控制】原則上原料血漿的檢測應(yīng)符合《中國藥典》及其他相關(guān)規(guī)定要求�����,小樣混合檢測時(shí)���,血漿混合的人份數(shù)量應(yīng)符合試劑盒/檢測方法的靈敏度要求。
【工藝設(shè)置和病毒風(fēng)險(xiǎn)評估】基于血液制品的安全性風(fēng)險(xiǎn)�,建議針對除人血白蛋白外的產(chǎn)品采用至少兩種以上作用機(jī)制互補(bǔ)的方式進(jìn)行病毒去除/滅活。持有人應(yīng)按照相關(guān)要求���,確定產(chǎn)品的有效病毒去除/滅活工藝步驟及工藝參數(shù)�����,鼓勵(lì)對產(chǎn)品制備的全工藝步驟(包括沉淀和層析步驟)的病毒清除能力進(jìn)行評估�����。鼓勵(lì)持有人在場地變更過程中合理優(yōu)化生產(chǎn)工藝(如增加層析步驟)和增設(shè)病毒去除/滅活步驟等�。
原則上��,進(jìn)行生產(chǎn)場地變更的首個(gè)階段應(yīng)遵循《血液制品去除/滅活病毒方法及驗(yàn)證指導(dǎo)原則》��,采用足以代表變更后商業(yè)工藝批次的樣品重新開展病毒滅活/去除驗(yàn)證;對于同類產(chǎn)品���,即對于工藝完全相同�、僅投料血漿不同的產(chǎn)品�����,在已有產(chǎn)品完成充分驗(yàn)證的前提下���,其他產(chǎn)品的驗(yàn)證可酌情簡化�����,但須有科學(xué)依據(jù)�。
驗(yàn)證原則上應(yīng)包括中國食品藥品檢定研究院病毒去除/滅活驗(yàn)證�����,該驗(yàn)證可僅對特定步驟開展�����。鼓勵(lì)持有人自行或委托第三方開展對全工藝病毒去除/滅活能力的評估及驗(yàn)證����。
在按照本指導(dǎo)原則開展驗(yàn)證的基礎(chǔ)上,持有人需進(jìn)一步對影響病毒去除/滅活效果的以下因素進(jìn)行重點(diǎn)評估, 作為累計(jì)風(fēng)險(xiǎn)的評估依據(jù),以加強(qiáng)場地轉(zhuǎn)移的風(fēng)險(xiǎn)控制和變更后產(chǎn)品的生產(chǎn)工藝過程控制。
①是否存在可能影響病毒清除效果的病毒去除/滅活工藝變更����,如,去除/滅活方法或工藝參數(shù)發(fā)生變化�;
②是否存在可能影響去除/滅活病毒效果的中間品質(zhì)量指標(biāo)檢測結(jié)果超出經(jīng)驗(yàn)證的參數(shù)范圍的情況(如pH、蛋白質(zhì)含量�����、純度�����、雜質(zhì)�����、保護(hù)劑種類及含量或其他可能干擾滅活過程的物質(zhì)含量等)�;
③用于病毒去除/滅活設(shè)備是否發(fā)生變更�,設(shè)備工作原理與變更前是否相同�����;
④既往產(chǎn)品病毒去除/滅活方法驗(yàn)證研究是否充分�。
持有人應(yīng)對病毒去除/滅活工藝、步驟�����、方法進(jìn)行全面評估��,除了需進(jìn)行全面的病毒去除/滅活效果驗(yàn)證外����,還應(yīng)特別關(guān)注該病毒去除/滅活方法對產(chǎn)品質(zhì)量和安全性��、有效性的影響。應(yīng)考慮滅活劑-蛋白等可能的相互作用�、工藝對目的蛋白的含量和完整性、結(jié)構(gòu)及臨床效果的影響�、是否會(huì)產(chǎn)生新的免疫原性����,以及可能引入的有毒殘留物風(fēng)險(xiǎn)等�����,建議開展活性成分的相關(guān)質(zhì)量屬性(如分子結(jié)構(gòu)�����、生物活性等)的比較研究,以確保產(chǎn)品質(zhì)量和安全性不受影響���。
如使用新的病毒去除/滅活方法,需參照上述要求證明該方法的科學(xué)�、有效����,未對產(chǎn)品質(zhì)量產(chǎn)生不良影響���,并經(jīng)充分驗(yàn)證。
對于場地變更后病毒去除/滅活工藝���,應(yīng)特別關(guān)注特定滅活步驟工藝參數(shù)的確定和設(shè)備的再驗(yàn)證����,包括對設(shè)備性能及溶液均一性進(jìn)行評估及驗(yàn)證�。應(yīng)提供病毒去除/滅活工藝條件(如制品的蛋白含量�����、pH���、離子強(qiáng)度����、加溫方式、溫度�、時(shí)間、載量����、過濾壓力等)與實(shí)際操作工藝參數(shù)對比表��,證明病毒去除/滅活工藝的代表性�。鼓勵(lì)對擬定的工藝參數(shù)范圍的病毒清除效果耐用性(病毒滅活/去除最差條件)開展對比研究和驗(yàn)證。
(3) 工藝驗(yàn)證
任何場地變更后�����,均應(yīng)參照GMP 相關(guān)條款及相關(guān)技術(shù)指南要求開展全面的設(shè)備驗(yàn)證�����,以確保用于生產(chǎn)的設(shè)備性能與變更前一致或更優(yōu)���。對于設(shè)備變更和/或規(guī)模放大的,則需開展設(shè)備變更對生產(chǎn)工藝參數(shù)(攪拌速度���、時(shí)間、溫度等)����、產(chǎn)品均勻度(如病毒滅活劑的分布等)及產(chǎn)品質(zhì)量影響的研究�����,確認(rèn)變更后工藝參數(shù)并開展全面的設(shè)備和工藝驗(yàn)證。如涉及凍干工藝�����,應(yīng)通過商業(yè)化批次生產(chǎn)驗(yàn)證凍干工藝參數(shù)��、凍干曲線等數(shù)據(jù)�,確證變更前后凍干工藝對產(chǎn)品質(zhì)量尤其是對生物活性等方面影響的可比性���。對于生產(chǎn)中直接接觸材料(如濾芯等)或容器(如一次性袋子等)變更����,需開展包材和容器的適用性(如可提取物/浸出物檢測)及產(chǎn)品質(zhì)量影響的研究,確認(rèn)變更后工藝參數(shù)并開展相關(guān)驗(yàn)證���。
鼓勵(lì)持有人利用場地變更的機(jī)會(huì)進(jìn)行生產(chǎn)工藝的升級優(yōu)化,如采用創(chuàng)新和改進(jìn)工藝(包括對乙醇分離工藝的改進(jìn))���,提高血漿利用率;使用層析的方法提高產(chǎn)品純度�����、去除有害殘留或有關(guān)物質(zhì)(如IgA�����、鋁離子等)和雜質(zhì)(如其他血液蛋白成分)�;使用不高于20nm 且能有效去除細(xì)小病毒的膜過濾器提高病毒去除能力等�。
鼓勵(lì)持有人加強(qiáng)生產(chǎn)過程控制�����,提升產(chǎn)品質(zhì)量標(biāo)準(zhǔn)�,如�����,增加特異性抗體��、IgA�����、鋁離子等檢測。
通常�,血液制品場地變更后應(yīng)至少進(jìn)行連續(xù)三批完整商業(yè)化規(guī)模工藝驗(yàn)證,鑒于血漿資源的稀缺性�,若工藝驗(yàn)證批次無法覆蓋最大商業(yè)化投漿規(guī)模,應(yīng)至少在動(dòng)態(tài)核查批次中包含最大商業(yè)化投漿規(guī)模批次����,并完成全項(xiàng)注冊檢驗(yàn)及可比性研究。對于制劑規(guī)模�����,企業(yè)可進(jìn)行風(fēng)險(xiǎn)評估確定工藝驗(yàn)證規(guī)模策略���。隨著血液綜合利用率提高�����,生產(chǎn)模式較為復(fù)雜�,需結(jié)合生產(chǎn)模式開展充分的中間產(chǎn)物穩(wěn)定性研究���。
【產(chǎn)品代表性】對于場地變更后工藝完全相同��、僅投料血漿不同的產(chǎn)品����,如人免疫球蛋白和某些特異性人免疫球蛋白,若某種代表性產(chǎn)品(通常為人免疫球蛋白)工藝驗(yàn)證充分����,在其他人免疫球蛋白品種申報(bào)時(shí),則可以用代表性產(chǎn)品工藝驗(yàn)證數(shù)據(jù)替代其驗(yàn)證工作���,但采用替代研究數(shù)據(jù)的人免疫球蛋白品種經(jīng)批準(zhǔn)后上市時(shí)��,前三批產(chǎn)品在批簽發(fā)時(shí)需進(jìn)行全項(xiàng)檢驗(yàn),并按照《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》開展相應(yīng)的穩(wěn)定性研究��。
持有人應(yīng)基于風(fēng)險(xiǎn)評估結(jié)果和平臺(tái)經(jīng)驗(yàn),制定合理工藝驗(yàn)證�、質(zhì)量和穩(wěn)定性研究方案���,可適當(dāng)減免驗(yàn)證批次,如血液制品同品 種僅為裝量不同的多規(guī)格情況���,可采用一個(gè)代表性規(guī)格連續(xù)三批�����、其他規(guī)格各一批的方式��,或括號法等進(jìn)行工藝驗(yàn)證��、質(zhì)量研究、檢驗(yàn)(包括注冊檢驗(yàn))和穩(wěn)定性考察�。對于濃度不同的情況(如 5%的白蛋白和20%的白蛋白,5%的靜丙和10%的靜丙等)��,可采用對每個(gè)規(guī)格各三批或括號法等進(jìn)行工藝驗(yàn)證�����、質(zhì)量研究(包括注冊檢驗(yàn))和穩(wěn)定性研究���。
3���、可比性
研究可比性研究是一個(gè)體系,包括研究設(shè)計(jì)�����、實(shí)施及對研究數(shù)據(jù)的評價(jià)等。持有人作為主體責(zé)任人�,應(yīng)基于風(fēng)險(xiǎn),根據(jù)產(chǎn)品特性和工藝特點(diǎn)合理�、規(guī)范、科學(xué)地進(jìn)行可比性研究設(shè)計(jì)�����。藥學(xué)可比性研究一般包括生產(chǎn)場地變更前后生產(chǎn)用原材料��、設(shè)備����、生產(chǎn)工藝及工藝參數(shù)�、過程控制��、產(chǎn)品質(zhì)量及穩(wěn)定性等的比較研究����,旨在證明場地變更后產(chǎn)品質(zhì)量不低于變更前,變更未對產(chǎn)品的安全性��、有效性和質(zhì)量可控性產(chǎn)生不良影響��。采用的比較方式包括與歷史數(shù)據(jù)的比較(如工藝可比性等)或平行試驗(yàn)比較(如質(zhì)量屬性可比性研究和穩(wěn)定性可比性等)。如果原液的變更可能會(huì)影響制劑�,則應(yīng)同時(shí)對原液和制劑進(jìn)行可比性研究。
工藝可比性研究中���,應(yīng)明確場地變更前后工藝變更的依據(jù)及目的,并以表格的形式提交兩個(gè)生產(chǎn)場地的設(shè)備�����、生產(chǎn)工藝����、工藝參數(shù)和產(chǎn)能等的對比�����。工藝可比性研究不僅僅需要對關(guān)鍵工藝參數(shù)及控制范圍進(jìn)行比較���,還應(yīng)對變更前后生產(chǎn)批次的實(shí)際工藝操作參數(shù)進(jìn)行比較,同時(shí)對于各步工藝的工藝性能(收率�����、雜質(zhì)去除率等)�����、中間品檢測結(jié)果(蛋白質(zhì)含量�����、純度等)進(jìn)行全面比較��。
質(zhì)量可比性研究中,除基于質(zhì)量標(biāo)準(zhǔn)對變更前后的多批次檢 定結(jié)果進(jìn)行比較外����,建議進(jìn)行變更前后產(chǎn)品拓展的質(zhì)量屬性研究�,并進(jìn)行比較���。此外,應(yīng)明確質(zhì)量分析項(xiàng)目���、分析方法(包括檢定場所)���、標(biāo)準(zhǔn)限度是否發(fā)生變更,質(zhì)量標(biāo)準(zhǔn)及質(zhì)控水平整體上不應(yīng)降低���。鼓勵(lì)采用更為靈敏的分析方法��,若由于分析方法升級導(dǎo)致質(zhì)量標(biāo)準(zhǔn)限度變更,則應(yīng)進(jìn)行充分的分析方法確證研究及方法橋接研究����,以表明變更后質(zhì)量標(biāo)準(zhǔn)并未實(shí)質(zhì)性降低���。檢定場所變更應(yīng)按照相關(guān)要求開展方法轉(zhuǎn)移研究。
穩(wěn)定性可比性研究中�����,在場地變更后有限的長期穩(wěn)定性研究中可能無法發(fā)現(xiàn)明顯差異��,加速和/或強(qiáng)制降解穩(wěn)定性研究可更直觀且快速地反映產(chǎn)品的降解模式、降解途徑和降解速率���,因此,除對場地變更前后多批次樣品進(jìn)行長期穩(wěn)定性可比性研究外�,還應(yīng)合理選擇敏感指標(biāo)�����,對強(qiáng)制降解�、加速穩(wěn)定性進(jìn)行可比性研究�,以明確變更前后降解趨勢���、降解速率等的可比性���。如果原液的變更可能會(huì)影響制劑的穩(wěn)定性����,則應(yīng)同時(shí)對原液(如涉及)和制劑進(jìn)行強(qiáng)制降解和/或加速穩(wěn)定性可比研究和長期穩(wěn)定性考察�。
4��、溝通交流
血液制品場地變更通常伴隨其他關(guān)聯(lián)變更,本指導(dǎo)原則無法就全部變更情況逐一列舉�,且變更分類往往需要結(jié)合研究結(jié)果�、產(chǎn)品知識等進(jìn)行風(fēng)險(xiǎn)評估和綜合判斷�����。鑒于血液制品的高風(fēng)險(xiǎn)性和稀缺性���,同時(shí)考慮到原料血漿的綜合利用度�����,鼓勵(lì)持有人按照《藥品上市后變更管理辦法(試行)》及生物制品變更技術(shù)指導(dǎo)原則的相關(guān)要求,通過溝通交流途徑����,就預(yù)期的已上市血液制品場地變更及關(guān)聯(lián)變更策略、變更類別����、支持變更的可比性方案和研究內(nèi)容、上市后變更管理方案等關(guān)鍵技術(shù)問題與相應(yīng)藥品監(jiān)管部門及技術(shù)單位進(jìn)行溝通����,特別是可能對血液制品質(zhì)量產(chǎn)生影響的重大變更。
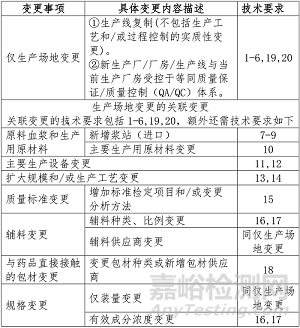
三�����、變更類型事項(xiàng)及技術(shù)要求
血液制品場地變更往往不是獨(dú)立發(fā)生����,可能伴隨或引發(fā)其他變更,即關(guān)聯(lián)變更�����。如,同時(shí)伴隨生產(chǎn)設(shè)備及生產(chǎn)工藝的變更���、處方中輔料變更�、質(zhì)量標(biāo)準(zhǔn)變更等�����。本指導(dǎo)原則對僅發(fā)生場地變更及其可能的關(guān)聯(lián)變更事項(xiàng)進(jìn)行例舉�����,并提出相關(guān)研究的技術(shù)要求�����。關(guān)聯(lián)變更需要在場地變更研究的基礎(chǔ)上�,參考各項(xiàng)變更要求分別開展研究工作��,并開展全面的變更可比性研究���。當(dāng)多個(gè)較低風(fēng)險(xiǎn)的變更事項(xiàng)關(guān)聯(lián)時(shí)�,可能導(dǎo)致整體變更風(fēng)險(xiǎn)升級��,建議關(guān)注多項(xiàng)關(guān)聯(lián)變更對藥品安全性��、有效性和質(zhì)量可控性產(chǎn)生的疊加影響�。具體分類原則參照上市后變更指導(dǎo)原則要求執(zhí)行,本指導(dǎo)原則僅以表格方式提出不同變更事項(xiàng)需要開展的技術(shù)研究考慮���。
由于各持有人實(shí)際情況不同�,存在多種變更情形�,本指導(dǎo)原則不能涵蓋所有關(guān)聯(lián)變更事項(xiàng)�����,對于本指導(dǎo)原則不涵蓋的變更事項(xiàng)�,持有人應(yīng)按照《藥品上市后變更管理辦法(試行)》及《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》,結(jié)合持有人實(shí)際情況開展相應(yīng)的可比性研究�。
技術(shù)要求:
1�、說明變更理由�。對生產(chǎn)場地變更情況具體描述�,包括生產(chǎn)廠的名稱(全稱)�����、地址(具體到廠房/車間、生產(chǎn)線�、地理位置等)和功能等。
2����、如涉及�,明確設(shè)備和生產(chǎn)原材料(級別�、檢定方法����、質(zhì)量標(biāo)準(zhǔn))、生產(chǎn)規(guī)模和工藝批量�、質(zhì)量標(biāo)準(zhǔn)(分析項(xiàng)目、方法和限度)����、與藥品直接接觸的包裝材料和容器等是否有改變,關(guān)注變更前后生產(chǎn)設(shè)施設(shè)備的性能、工作原理��、生產(chǎn)能力等與生產(chǎn)工藝的匹配性���。如涉及���,提供支持性資料。
3�����、至少進(jìn)行連續(xù)三批商業(yè)生產(chǎn)規(guī)模原液、半成品和制劑生產(chǎn)工藝驗(yàn)證(若涉及多個(gè)規(guī)格,且僅灌裝量不同���,應(yīng)基于風(fēng)險(xiǎn)評估結(jié)果和平臺(tái)經(jīng)驗(yàn)制定工藝驗(yàn)證方案�����,可適當(dāng)減免驗(yàn)證批次�����,如采用一個(gè)代表性規(guī)格的進(jìn)行連續(xù)三批、其他規(guī)格各一批或括號法等進(jìn)行工藝驗(yàn)證)��。若工藝驗(yàn)證批次無法覆蓋最大商業(yè)化投漿規(guī)模�,應(yīng)至少在動(dòng)態(tài)核查批次中包含最大商業(yè)化投漿規(guī)模批次�,并完成全項(xiàng)注冊檢驗(yàn)及可比性研究。應(yīng)明確驗(yàn)證批次規(guī)模(是否與設(shè)計(jì)生產(chǎn)能力相符)����、生產(chǎn)工藝代表性的分析(如,是否可覆蓋常規(guī)生產(chǎn)規(guī)模范圍)�。工藝驗(yàn)證應(yīng)包括對連續(xù)生產(chǎn)批次符合其預(yù)定過程控制標(biāo)準(zhǔn)及質(zhì)量標(biāo)準(zhǔn)進(jìn)行的分析�����;對無菌生產(chǎn)�����、滅菌工藝的驗(yàn)證�����;工藝對產(chǎn)品相關(guān)雜質(zhì)種類和含量影響的分析驗(yàn)證��;對工藝相關(guān)雜質(zhì)的去除能力驗(yàn)證;委托中檢院開展病毒滅活/去除效果驗(yàn)證�;必要時(shí)��,驗(yàn)證內(nèi)容還可能涉及中間產(chǎn)物貯藏時(shí)間的驗(yàn)證,超濾膜和層析介質(zhì)使用壽命的研究等���。
4���、提供變更可比性研究方案及研究結(jié)果�。除特殊要求外�����,應(yīng)對至少三批變更后原液��、半成品和制劑進(jìn)行質(zhì)量分析���,研究(鑒別�、生物活性���、純度��、雜質(zhì)��、污染等)����,并與變更前數(shù)據(jù)(充分的歷史批次)進(jìn)行可比性研究�。若涉及�����,檢測場地發(fā)生變更的,需提供支持檢測方法學(xué)轉(zhuǎn)移的技術(shù)資料���。
5、除特殊要求外�����,提供變更前后原液(如涉及)和制劑加速和/或降解條件下的結(jié)果�。提供變更前后原液(如涉及)和制劑的實(shí)時(shí)/實(shí)際條件下的穩(wěn)定性研究數(shù)據(jù)。對變更前后的原液(如涉及)和制劑的加速和/或強(qiáng)制降解以及實(shí)時(shí)/實(shí)際條件下的穩(wěn)定性進(jìn)行可比性研究���。變更前的數(shù)據(jù)可為歷史穩(wěn)定性檢定結(jié)果。若涉及����,應(yīng)進(jìn)行運(yùn)輸穩(wěn)定性研究�。
6�����、制定穩(wěn)定性研究方案����。繼續(xù)進(jìn)行批準(zhǔn)后長期穩(wěn)定性研究,以確證原液(如涉及)和制劑的放置時(shí)間/有效期�����。承諾報(bào)告長期穩(wěn)定性研究中出現(xiàn)的不合格情況��。
7���、原料血漿不應(yīng)從血液傳染病高危人群中收集。提供所屬采漿機(jī)構(gòu)至少近6 個(gè)月的固定獻(xiàn)血漿者的陽性檢測率(根據(jù)獻(xiàn)血漿者例數(shù)和捐獻(xiàn)血漿次數(shù))和新獻(xiàn)血漿者的陽性檢測率���。提供原料血漿復(fù)核檢驗(yàn)質(zhì)量回顧及風(fēng)險(xiǎn)評估資料。
8、具備采漿機(jī)構(gòu)完整資料和資質(zhì)證明�����。具有持有人與采漿機(jī)構(gòu)的合同(質(zhì)量協(xié)議等)文件�����。境外生產(chǎn)產(chǎn)品應(yīng)具備原料血漿來源于非瘋牛病疫情地區(qū)/國家的證明�。原料血漿的采集�、檢驗(yàn)、貯藏和運(yùn)輸應(yīng)當(dāng)符合相關(guān)規(guī)定�。
9����、應(yīng)當(dāng)與單采血漿站建立信息交換系統(tǒng)���,出現(xiàn)情況及時(shí)交換信息�。應(yīng)當(dāng)建立原料血漿的追溯系統(tǒng)�����,確保每份原料血漿可追溯至獻(xiàn)血漿者�,并可向前追溯到獻(xiàn)血漿者最后一次采集的血漿之 前至少檢疫期內(nèi)所采集的原料血漿�����,或該獻(xiàn)血漿者的血液標(biāo)本��。該獻(xiàn)血漿者在某一次獻(xiàn)血漿后不再獻(xiàn)血漿����,可用其血液標(biāo)本檢定,合格時(shí)才能放行檢疫期前的原料血漿。
10�����、說明原材料變更理由�。生產(chǎn)用原材料應(yīng)滿足生產(chǎn)需求,且符合《中國藥典》及相關(guān)指導(dǎo)原則規(guī)定�����。提供變更后生產(chǎn)用原材料的來源����、質(zhì)量標(biāo)準(zhǔn)、檢定報(bào)告等資料�����。對于變更來源的�,變更后級別和質(zhì)量應(yīng)不低于變更前。生產(chǎn)過程中有機(jī)溶劑的使用及殘留限值的規(guī)定應(yīng)嚴(yán)格按照《中國藥典》�、ICH“殘留溶劑測定法”的規(guī)定,避免使用第一類溶劑�����,限制使用第二類溶劑。如采用有機(jī)溶劑或其他物質(zhì)進(jìn)行提取���、純化或滅活處理等��,后續(xù)純化工藝應(yīng)保證可有效去除制品中的有機(jī)溶劑或其他物質(zhì)���,去除工藝應(yīng)經(jīng)驗(yàn)證;如涉及�,修訂原液/制劑質(zhì)量標(biāo)準(zhǔn),設(shè)定殘留物的檢定項(xiàng)�,并對新方法進(jìn)行方法學(xué)驗(yàn)證。對變更后生產(chǎn)用原材料進(jìn)行安全風(fēng)險(xiǎn)評估���,除原料血漿外,原則上不使用人源或動(dòng)物源性材料�;如涉及,應(yīng)提供充分的依據(jù)����,并提供生產(chǎn)原料不存在 BSE/TSE 潛在風(fēng)險(xiǎn)的信息和證據(jù),如供應(yīng)商名稱�、原料來源的種屬和組織、動(dòng)物的原產(chǎn)地���、使用情況���。對于自制的生產(chǎn)用原材料(如層析填料等)���,應(yīng)提供其全套生產(chǎn)、質(zhì)量��、穩(wěn)定性等研究資料����。
11、說明變更理由���。明確擬變更設(shè)備的信息�����,進(jìn)行變更前后設(shè)備操作原理和關(guān)鍵技術(shù)參數(shù)的相似點(diǎn)和差異性的對比�����。對新增設(shè)備開展運(yùn)行確認(rèn)(OQ)和性能確認(rèn)(PQ)���。如涉及干熱滅活�����,則須重新開展對變更后產(chǎn)品干熱步驟(含凍干)的病毒滅活驗(yàn)證��。
12��、如涉及��,一次性使用系統(tǒng)應(yīng)具有供應(yīng)商質(zhì)量保證/質(zhì)量體系和核心驗(yàn)證文件�����。持有人應(yīng)結(jié)合血液制品生產(chǎn)對一次性使用系統(tǒng)進(jìn)行驗(yàn)證�����,包括化學(xué)兼容性��、吸附能力�、細(xì)菌挑戰(zhàn)�����、顆粒物�����、可提取物和/或浸出物等方面��。若可提取物和/或浸出物的種類及含量較變更前發(fā)生變化時(shí)�����,應(yīng)進(jìn)一步評估這種變化對生產(chǎn)工藝(包括下游工藝)及產(chǎn)品的影響�����,研究新出現(xiàn)的可提取物和/或浸出物是否會(huì)與藥物發(fā)生相互作用��。
13�����、說明變更理由���。詳細(xì)說明變更的原因及具體變更情況(工藝路線�����、生產(chǎn)過程控制���、可接受范圍等)����,進(jìn)行變更前后工藝及過程控制的對比��。對生產(chǎn)工藝的變更應(yīng)進(jìn)行全面的研究���,根據(jù)風(fēng)險(xiǎn)評估分析對原液和制劑質(zhì)量產(chǎn)生的影響�。
14�、提供生產(chǎn)工藝流程圖,標(biāo)明工藝步驟和過程控制參數(shù)����,顯示材料加入環(huán)節(jié)。對擬定生產(chǎn)工藝簡要敘述�����。明確變更后生產(chǎn)工藝規(guī)模����、流程����、工藝和過程參數(shù)(如層析介質(zhì)����、緩沖液�、洗脫液、流速����、填料載量、收峰條件等)�����、過程控制要求(如回收率����、純度和雜質(zhì)、微生物污染監(jiān)測等)����。
15、說明變更理由�����。提供變更前后質(zhì)量標(biāo)準(zhǔn)對比表。提供變更前后多批次質(zhì)量可比性研究數(shù)據(jù)����,以證明變更后質(zhì)量不低于變更前。對于分析方法變更的�,須提供變更后方法學(xué)驗(yàn)證資料及變更前后方法的可比性研究資料。增加標(biāo)準(zhǔn)檢定項(xiàng)目和/或變更分析方法變更的�����,需進(jìn)行中檢院單項(xiàng)標(biāo)準(zhǔn)復(fù)核及檢驗(yàn)���。
16�、說明變更理由���。提供充分的處方開發(fā)研究資料和變更前后制劑處方對比表�。如新增輔料����,應(yīng)提供詳細(xì)的證明性材料(來源、質(zhì)量標(biāo)準(zhǔn)和/或內(nèi)控標(biāo)準(zhǔn)�、檢驗(yàn)報(bào)告),輔料的關(guān)聯(lián)審評信息或無需關(guān)聯(lián)審評說明。原則上制劑的輔料應(yīng)符合“生物制品生產(chǎn)用原材料及輔料的質(zhì)量控制規(guī)程”的相關(guān)規(guī)定����。如涉及���,對質(zhì)量標(biāo)準(zhǔn)進(jìn)行必要的修訂�。
17�����、必要時(shí)應(yīng)根據(jù)產(chǎn)品的特點(diǎn)���,選擇進(jìn)行動(dòng)物安全性���、有效性評估,或臨床研究��。
18���、說明變更理由�。闡述包材的選擇依據(jù)���、合理性和適用性信息��。提供變更前后包材對比表�����。提供變更后包材的來源����、質(zhì)量標(biāo)準(zhǔn)、檢定報(bào)告���、登記信息�����、包材相容性和容器密封完整性等資料���。預(yù)充式注射器需開展滑動(dòng)力等研究,應(yīng)提供其檢測方法及方法學(xué)驗(yàn)證資料�����,并納入質(zhì)量標(biāo)準(zhǔn)進(jìn)行控制�����。
19、當(dāng)藥學(xué)可比性研究數(shù)據(jù)不足以支持變更可比性時(shí)�,應(yīng)進(jìn)行非臨床和/或臨床的橋接研究來確認(rèn)可比性。否則需提供免除的依據(jù)���。
20�����、必要時(shí),進(jìn)行特殊安全性試驗(yàn)(涉及過敏����、溶血和血管刺激等)。
四�、其他考量
1、增加生產(chǎn)場地/生產(chǎn)線
增加生產(chǎn)場地或現(xiàn)有生產(chǎn)車間增加生產(chǎn)線原則上應(yīng)符合本指導(dǎo)原則的要求�。持有人應(yīng)在持續(xù)生產(chǎn)過程中確保不同場地產(chǎn)品的質(zhì)量可比性。
2����、非臨床研究及臨床試驗(yàn)
血液制品場地變更后產(chǎn)品,非臨床研究和/或臨床試驗(yàn)的方式和程度應(yīng)結(jié)合藥學(xué)可比性研究的結(jié)果���、對產(chǎn)品性質(zhì)了解的知識水平�����、已完成的相關(guān)非臨床和/或臨床試驗(yàn)數(shù)據(jù)以及該產(chǎn)品的臨床應(yīng)用等情形�,基于具體問題具體分析的原則來確定。非臨床研究方面����,原則上應(yīng)完成制劑安全性研究(局部刺激試驗(yàn)及溶血試驗(yàn)等)。臨床試驗(yàn)方面�,應(yīng)充分評估變更后產(chǎn)品的質(zhì)量屬性對臨床安全性和有效性的影響,當(dāng)特定質(zhì)量屬性與安全性和有效性之間的關(guān)系尚未確定��,或變更前后產(chǎn)品的質(zhì)量屬性存在差異的情況下����,應(yīng)開展相應(yīng)的臨床試驗(yàn)。
3���、風(fēng)險(xiǎn)管控
參考ICHQ12 等先進(jìn)監(jiān)管模式�,持有人可以自主選擇使用變更管理工具����,如既定條件�����、上市后變更管理方案(PACMP)和生命周期管理等�����,實(shí)現(xiàn)對血液制品上市后變更策略性的規(guī)劃和高效的管理�����。
五���、參考文獻(xiàn)
1�、WHO. WHO EXPERT COMMITTEE ON BIOLOGICAL STANDARDIZATION (52nd report: WHO TRS N°924 (A4): 2001).Annex 4: Guidelines on viral inactivation and removal procedures intended to assure the viral safety of human blood plasma products.
2、國家藥典委員會(huì).中華人民共和國家藥典[S].2020 版.北京:中國醫(yī)藥科技出社版.
3�����、NMPA.血液制品去除/滅活病毒技術(shù)方法及驗(yàn)證指導(dǎo)原則(2002).
4���、NMPA. 已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)(2021).
5�����、NMPA. 已上市疫苗藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)(2024).
6�、NMPA.疫苗生產(chǎn)場地變更質(zhì)量可比性研究技術(shù)指導(dǎo)原則(2014).
7、ICH Q5C: Stability testing of biotechnological /biological products. November 1995.