2022.12.9�����,F(xiàn)DA發(fā)布“醫(yī)療器械上市前遞交材料中人為因素信息的內(nèi)容”的指南草案,以修訂2016年發(fā)布的“人為因素審查最高優(yōu)先級器械清單”指南草案�。


該草案征求意見截止日期為2023.3.9���。
該指南草案提供了一個基于風險的框架�����,以指導制造商和FDA工作人員關于人為因素的信息,這些信息應包括在上市前遞交文檔中,以提高FDA審查過程的效率���。
【草案背景】
醫(yī)療器械的一個獨特方面是器械-用戶界面交互對其安全使用的關鍵作用�����。
在設備開發(fā)過程中�,制造商通常會對人機界面進行人為因素評估�����。
2016年2月3日,F(xiàn)DA在《聯(lián)邦公報》中公布了“人為因素審查最高優(yōu)先級器械清單”指南草案���。在考慮了利益相關方對2016年2月3日發(fā)布的指南草案的反饋后,F(xiàn)DA發(fā)布了一份修訂版指南草案,現(xiàn)標題改為“醫(yī)療器械上市申請中人為因素信息的內(nèi)容”���。該指南草案提供了FDA關于提交人為因素信息的基于風險的政策,用于上市前審查,以回應利益相關者的反饋�����。
定稿后,該指南草案將用于補充FDA指南“將人為因素和可用性工程應用于醫(yī)療器械(以下簡稱為“人為因素指南”)�����。最終定稿后�����,F(xiàn)DA打算同時修訂“人為因素指南”�,主要修訂包括如下四點:
1. 將“人為因素指南”第3節(jié)中的定義替換為本指南中包含的定義;
2. 修訂“人為因素指南”第9節(jié)“文件”���;
3. 將“人為因素指南”附錄A“人為因素和可用性工程報告”替換為本指南第五節(jié)的交叉引用�����;
4. 酌情對“人為因素指南”進行任何其他修訂�����。
FDA認識到并預計該機構(gòu)和行業(yè)可能需要多達60天的時間來開展活動���,以實施本指南中的政策。如果在最終指南發(fā)布前或發(fā)布后60天內(nèi)�,F(xiàn)DA收到的上市申請中沒有關于上市申請中人為因素信息內(nèi)容的新信息�����,CDRH工作人員一般不打算在審查申請時要求提供此類信息�����。然而���,如果收到任何此類信息,CDRH確實打算進行審查。
FDA認識到并預計FDA和行業(yè)可能需要多達60天的時間來開展活動�,以實施本指南中的政策���。
如果在最終指南發(fā)布后60天內(nèi),F(xiàn)DA收到的上市申請中沒有關于上市申請中人為因素信息內(nèi)容的新信息�����,CDRH工作人員一般不打算在審查申請時要求提供此類信息�。然而�����,如果收到任何此類信息�,CDRH確實打算進行審查。
【人為因素評估的目標】
確保器械用戶界面的設計能夠消除或盡可能減少器械使用過程中可能導致傷害或降低醫(yī)療質(zhì)量的使用錯誤���。
在基于風險的人為因素評估方法中需要考慮的主要因素���,包括識別已有或修改的關鍵任務,以及消除或減少與使用相關的危險�。
【FDA目前對可用性信息的審核】
FDA關于器械上市前遞交的決策是基于適用的法定和監(jiān)管標準的安全性和有效性的合理保證���。就相關程度而言�,人為因素僅構(gòu)成FDA評估的組成部分之一�����。
雖然FDA認為最大限度地降低使用相關的風險是最理想的�,但消除所有使用相關的器械風險可能是不必要的�,也是不實際的�。
在適當?shù)那闆r下�����,上市前遞交信息應證明在器械設計中考慮了預期用戶的需求�����,并且器械對于預期用戶�����、用途和使用環(huán)境是安全有效的���。
因此�����,在適當?shù)那闆r下�����,上市任務應包括解釋關鍵任務存在與否的信息�、風險緩解策略的驗證測試以及剩余風險的描述�。包括適當?shù)娜藶橐蛩匦畔⒖梢酝ㄟ^減少對AI問題來提高FDA審查的效率���。
【適用范圍】
本指南旨在幫助提交者和FDA工作人員確定哪些人為因素評估信息應包含在醫(yī)療器械的上市申請中�,包括510(k)s�、De Novo���、PMA和HDE申請。
【可用性遞交類別】
將人為因素工程信息包括在上市前遞交文件中的目的是�,通過證明器械的用戶界面適合預期用戶、用途和使用環(huán)境���,幫助制造商滿足適用的法律標準���。
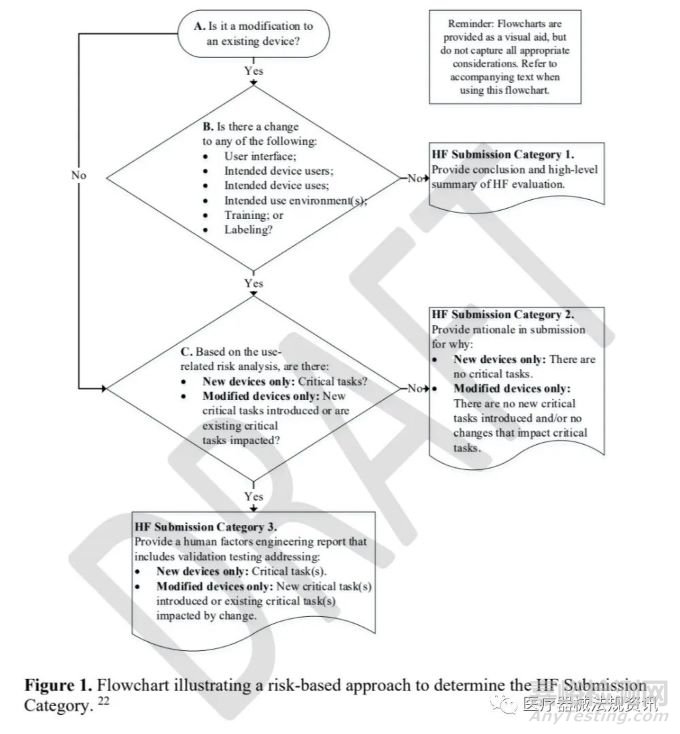
本草案中使用流程圖、表格和文本來指導提交者通過基于風險的方法推薦提交者應在其上市前遞交中包括哪些人為因素工程信息�。FDA將這種基于風險的方法稱為人為因素(HF)遞交類別。提交者應使用圖1中的流程圖�,并使用其配套文本來回答每個決策點提出的問題。確定哪種HF遞交類別適合支持其上市前遞交�����。
該流程圖基于新器械和FDA已授予上市許可的器械的使用適應癥和使用相關風險分析���。FDA基于關鍵任務的存在或修改進行HF遞交分類,考慮技術特征或使用說明的變化�����,如果相關。提交人應使用下述使用相關風險分析和決策點來幫助確定其上市提交的HF提交分類�。提交者在使用圖1確定其遞交屬于哪一類HF遞交后,還應參考表1提交FDA推薦的人為因素工程信息�����。
通過上圖的流程���,可將人為因素(HF)遞交類別分為:
• HF Submission Category 1
可用性遞交類別1
• HF Submission Category 2
可用性遞交類別2
• HF Submission Category 3
可用性遞交類別3
【可用性遞交信息】
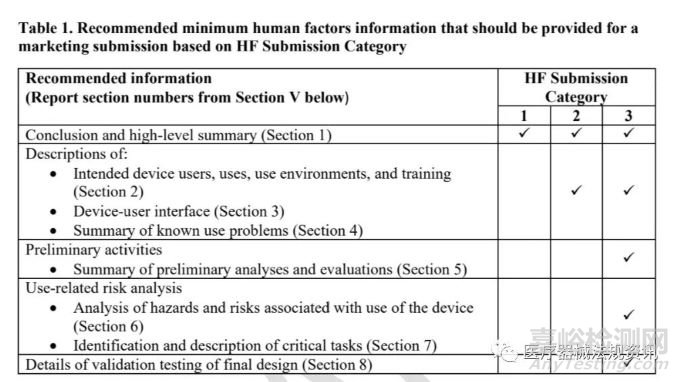
• 對于HF遞交類別1:提供HF評估的結(jié)論和高級摘要:
提交的文件應包括一份聲明,證明器械的修改不會影響修改器械的人為因素考慮�,并利用(如果適用)先前的人為因素工程評估來提供結(jié)論和高級摘要。對于屬于HF提交類別1的器械���,建議提交內(nèi)容見表1�。
• 對于HF提交類別2:提交理由:沒有關鍵的任務(僅限新器械)�����;或者沒有引入新的關鍵任務和/或沒有影響關鍵任務的變化(僅限于修改后的器械):
提交人應提交一份理由�����,清楚地描述他們卻確定新器械沒有關鍵任務,或修改后的器械沒有影響關鍵任務的依據(jù)�。這個基本原理應該基于第四節(jié)中提到的決策。關于屬于HF提交類別2的器械的建議提交內(nèi)容���,請參見表1。
?
• 對于HF遞交類別3:提供一份人因工程報告���,包括驗證測試�,解決:
* 關鍵任務(僅限新器械�����;見表2)�����;或
* 新的關鍵任務引入或現(xiàn)有的關鍵任務受變更影響(僅修改器械�;見表3):
應在HF提交類別3中向FDA提交一份全面的人為因素工程報告���,該報告包括本指南第IV節(jié)中描述的人為因素工程報告的所有要素。請注意,如果關鍵任務受到改良器械的影響�����,但現(xiàn)有的風險控制措施仍然可以接受�����,應在提交中提供理由���,作為人為因素信息的一部分。
?
【人為因素信息的建議內(nèi)容】
上市前遞交制造商的風險管理���、人為因素工程測試(如適用)和設計優(yōu)化過程的內(nèi)部文件有助于提供證據(jù)���,在適當?shù)那闆r下,證明在設計中考慮了預期用戶的需求���,且器械對于預期用戶���、用途和使用環(huán)境是安全有效的�����。
FDA建議人為因素信息應由制造商保存,不管是否提交給FDA���。制造商必須在適用法律要求的范圍內(nèi)保存記錄���。
人為因素工程信息應總結(jié)所執(zhí)行的評估。這種信息通常不包括來自人為因素確認測試的所有原始數(shù)據(jù)���。
作為人因工程過程一部分的文件或分析應包含在上市前提交中提供的人因工程信息中�。這包括風險分析���,側(cè)重于用戶與器械的交互和特定的風險分析�����、過程���、結(jié)果和結(jié)論。
人為因素工程信息的推薦結(jié)構(gòu)如下:
• 第1部分: 結(jié)論和高級總結(jié)
提交者應該從結(jié)論開始�����,說明器械的用戶界面是否被認為是為預期用戶、用途和使用環(huán)境充分設計的�,以及是否進行了新的人為因素測試來支持這一結(jié)論。FDA建議提交者從人因工程評估(例如�����,使用相關風險)的高級摘要開始�,包括進行評估的基本原理���,以及進行的人因工程過程的摘要(例如���,人因工程分析和評估,器械-用戶界面修改和驗證測試)和結(jié)果的分析���。適用時�,本節(jié)應討論人為因素確認試驗后任何剩余的使用相關風險�。提交者應說明為什么進一步降低風險不可行,這是基于對器械的效益風險分析���。
• 第2部分:目標器械用戶�、用途���、使用環(huán)境和培訓的描述
應包括:
1. 目標用戶群的描述
如果有一個以上不同的用戶群體�,應描述每個群體。描述應包括用戶群之間在能力或使用責任方面有意義的差異�����,這些差異可能會影響他們與器械的交互�����。這包括可能使用同一器械執(zhí)行不同任務的外行和醫(yī)療保健專業(yè)用戶���,或者可能在器械上執(zhí)行不同任務的不同類型的專業(yè)人員�。
2. 器械預期用途的概述
3. 器械使用的操作環(huán)境和設備操作的關鍵方面的總結(jié)���,包括:
* 在使用器械之前�,用戶是否應該或必須接受醫(yī)療專業(yè)人員的培訓
* 如何在臨床應用中使用該器械
* 設置���、維護���、清理和再處理信息
4. 預期使用環(huán)境(例如醫(yī)院、醫(yī)療救護車�����、家用)以及可能影響用戶與設備交互的環(huán)境特征(例如強光、振動�����、環(huán)境噪音���、高水平活動)的概述
5. 用戶將接受的任何培訓的描述
可以附上培訓材料的樣品,如視頻�、演示幻燈片或小冊子。
• 第3部分:設備-用戶界面的描述
如果適用���,應包括:
1. 器械及其用戶界面的圖形表示(如照片�、插圖���、線條畫)
這應描述整個器械和用戶將與之交互的用戶界面的所有組件(例如���,顯示和功能屏幕、報警揚聲器�、控制裝置、鍵盤�����、專用按鈕、門�、要連接的組件、固定夾)�。
2. 器械用戶界面的書面描述
將隨器械提供給用戶的標簽副本(例如,使用適應癥�、用戶手冊、快速入門指南���、包裝)�。
3. 器械操作順序和用戶預期
4. 與用戶界面交互的概述
這應包括為使用該器械而執(zhí)行的用戶動作的序列�,以及在適當時產(chǎn)生的器械響應;
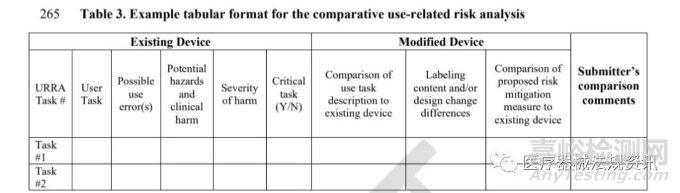
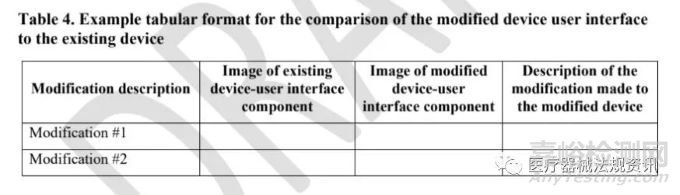
5. 對于改良的器械���,考慮提供申請器械和現(xiàn)有器械的比較信息(格式示例見表4)���。
?
?
•第4部分: 已知使用問題概述
應描述相同器械(如適用)或相似類型器械之前型號的所有已知使用問題。
FDA建議提交者聲明沒有已知的使用問題�����,如果適用���。對于專門針對現(xiàn)場使用問題進行修改的器械�,應討論這些問題和器械修改。
• 第5部分: 初步分析和評估總結(jié)
應確定所使用的初步分析和評估方法(例如�,特定的分析技術、形成性評估)�����,總結(jié)這些分析和評估的關鍵結(jié)果�����,描述相應的用戶界面設計的修改�����,并討論構(gòu)成人為因素驗證試驗方案開發(fā)的關鍵發(fā)現(xiàn)�。
• 第6部分: 與器械使用相關的危險和風險分析
應包括與使用相關的風險分析文件和/或比較任務分析�����,如適用�。這是綜合風險分析的典型摘錄,包含通過初步分析和評估確定的所有與使用相關的危險和風險�����,包括與潛在使用錯誤相關的危險和風險。
使用相關風險分析文件應是一份不斷更新的文件�����;在整個器械設計過程中�,應對已識別的風險和危害進行更新。
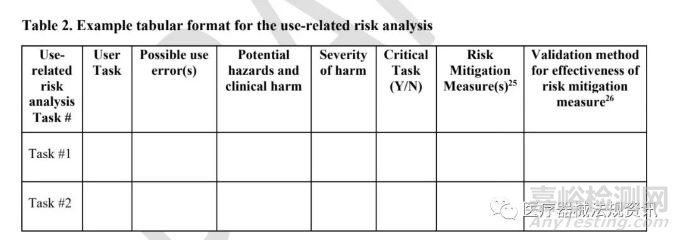
FDA認為以表格形式組織信息是有用的�����。表2提供了一個表格格式的例子�。本示例提供了評估與您的設備相關的使用相關風險的最低建議信息。對于HF遞交類別3中的改裝器械�,提交者應提供對比任務分析(見表3中的表格格式示例),將改裝器械使用相關風險分析與現(xiàn)有器械使用相關風險分析進行比較�。如果確定導致任務、相關危害���、和/或風險緩解措施修改的器械變更不需要新的HF驗證試驗數(shù)據(jù)來支持器械的使用安全性�,請?zhí)峁├碛伞?/span>
• 第7部分:關鍵任務的識別和描述
1.應解釋根據(jù)使用相關風險分析文件識別關鍵任務的過程
由于關鍵任務是由潛在危險的嚴重程度決定的�����,F(xiàn)DA建議提交者描述正在使用的嚴重程度,并在適當?shù)臅r候用作參考�。例如,如果提交者使用自愿共識標準中的定性五級嚴重性評級�,則應包括一個嚴重性級別表,描述每個級別并引用適用的標準�。
2. 列出并描述關鍵任務
對于HF提交類別3,如果相關的和合理的任務不需要新的HF驗證測試數(shù)據(jù)來支持器械的使用安全���,則提交者應提供單獨的表格���,突出顯示新的關鍵任務。提交人還應描述人為因素驗證測試中包含的每個使用場景�����,并列出構(gòu)成每個使用場景的關鍵和非關鍵任務���。
當修改現(xiàn)有器械時,F(xiàn)DA建議提交者將新器械用戶界面與他們自己的現(xiàn)有器械進行比較�。FDA建議以表格形式完成這一比較。表格格式示例如表4所示�。
除了整個器械的使用相關風險分析文件外,提交者還應包括使用相關風險分析的子集。
FDA建議包括修改過的器械用戶界面組件的照片�,包括對標簽的修改,如手冊中的警告聲明�����。
提交者應該列出受修改影響的任何關鍵任務���。提交者還應討論與修改相關的風險是否可接受�����,并評估提議的更改是否保證人為因素確認測試�����。
• 第8部分: 最終設計的HF驗證測試的細節(jié)
應該總結(jié)所有進行的HF驗證活動�����。除了測試結(jié)果之外���,這一部分還應包含對所有使用錯誤和問題的綜合分析,這些錯誤和問題可能會導致現(xiàn)實世界使用中的傷害�����,還應包含針對測試結(jié)果對用戶界面進行的所有設計修改的描述,以及收益風險討論�。
應附上完整的測試方案和測試中使用的所有腳本和表格的樣本。
提交者應提供剩余風險分析和現(xiàn)有緩解控制措施可接受的理由�。盡管消除所有剩余風險可能不切實際,但提交者應具備對使用錯誤和使用相關風險緩解措施進行系統(tǒng)分析的證據(jù)���。
當確定剩余風險不可接受時�,提交者應評估風險控制和緩解措施�,以確定降低風險的其他方法。
草案文末提供了3個假設的場景示例
旨在使用圖1中的流程圖及其配套文字說明FDA基于風險的方法來確定HF遞交類別���?����;贖F遞交類別�����,F(xiàn)DA推薦的支持上市提交的HF信息在每種情況下都有概述�����。這些例子并沒有說明每種遞交類型�,也沒有說明可能適合每種情況的人為因素信息�。此外,描述對現(xiàn)有器械進行修改的示例是基于一個假設���,即制造商已經(jīng)決定它需要提交一份新的上市前遞交材料�����。因此�����,這些示例并不是為了解釋何時需要提交新的上市前遞交材料�。此外�����,這些示例并不旨在全面代表新器械或現(xiàn)有器械改進的上市子任務中應包含的內(nèi)容�。