摘 要 Abstract
我國(guó)確立了基于風(fēng)險(xiǎn)啟動(dòng)藥品注冊(cè)核查檢驗(yàn)的模式�,2021 年12 月國(guó)家藥品監(jiān)督管理局藥品審評(píng)中心發(fā)布《藥品注冊(cè)核查檢驗(yàn)啟動(dòng)工作程序(試行)》,進(jìn)一步明確核查檢驗(yàn)具體程序�。本文從監(jiān)管要求、外部協(xié)作管理及申辦者內(nèi)控制度三個(gè)維度出發(fā)�,結(jié)合研究機(jī)構(gòu)/ 研究者合規(guī)、第三方服務(wù)機(jī)構(gòu)合規(guī)�、臨床數(shù)據(jù)合規(guī)及人類遺傳資源合規(guī),探討臨床研究中的合規(guī)難題�,以期為我國(guó)醫(yī)藥企業(yè)臨床研究合規(guī)管理提供借鑒。
China initiated a risk-based model on examination and inspection of drug registration. Tentative Procedures on Examination and Inspection of Drug Registration promulgated by Centre for Drug Evaluation ("CDE") under National Medical Products Administration ("NMPA") in December 2021 intended to standardize detailed procedures on examination and inspection of drug registration. This paper aims to probe into critical compliance issues in clinical studies from the perspectives of regulatory requirement, external cooperation management and sponsor's internal control system by highlighting some key compliance considerations for investigators / institutions, third-party service providers, clinical data security and human genetic resources, in an effort to shed some light on compliance management of clinical studies by biotech, biopharma and pharmaceutical companies.
關(guān)鍵詞 Key words
臨床研究�;合規(guī)管理�;數(shù)據(jù)合規(guī)�;人類遺傳資源管理;合規(guī)體系建設(shè)
clinical study; compliance management; data compliance; human genetic resources management; compliance management systems
臨床研究分工細(xì)致�、法律關(guān)系復(fù)雜。申辦者(指負(fù)責(zé)臨床研究的發(fā)起�、管理和提供臨床研究經(jīng)費(fèi)的個(gè)人、組織或者機(jī)構(gòu))作為臨床研究質(zhì)量最終責(zé)任人面臨越來越多的合規(guī)難題�。申辦者應(yīng)如何建立臨床研究合規(guī)管理體系?如何妥善管理各臨床研究參與方�?如何開展臨床數(shù)據(jù)合規(guī)管理?如何精準(zhǔn)把握人類遺傳資源監(jiān)管要求�?本文將針對(duì)以上合規(guī)難題,結(jié)合境外臨床研究合規(guī)實(shí)踐�,探討相關(guān)解決路徑,以期為我國(guó)醫(yī)藥企業(yè)臨床研究合規(guī)管理提供借鑒�。
一、臨床合規(guī)管理體系
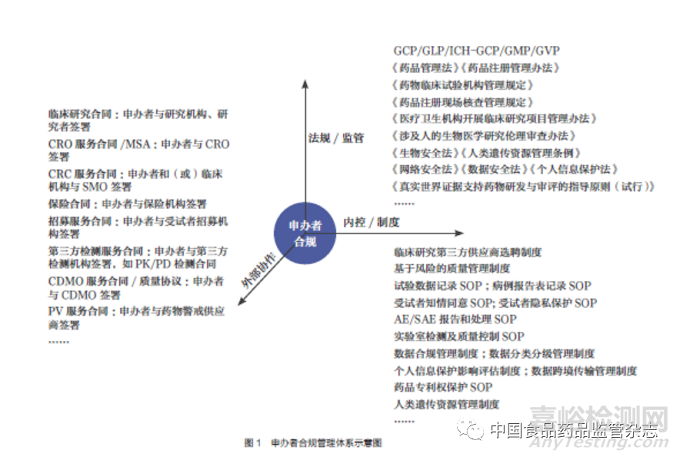
申辦者作為臨床研究數(shù)據(jù)質(zhì)量和可靠性的最終責(zé)任人[1]�,需要在遵循監(jiān)管要求的基礎(chǔ)上,充分協(xié)調(diào)并管理外部協(xié)作資源�,不斷建立并完善內(nèi)控制度�。筆者建議可從三個(gè)維度對(duì)臨床研究進(jìn)行合規(guī)管理,以完善合規(guī)管理體系�,如圖1 所示。
(一)監(jiān)管要求
《藥品管理法》《藥品注冊(cè)管理辦法》《藥物臨床試驗(yàn)質(zhì)量管理規(guī)范》(GCP)等法規(guī)是我國(guó)臨床研究監(jiān)管的主要依據(jù)�,立法和監(jiān)管部門還就研究機(jī)構(gòu)、倫理審查、數(shù)據(jù)核查與數(shù)據(jù)安全�、人類遺傳資源管理等發(fā)布了相關(guān)法律法規(guī)和規(guī)范性文件,并開展基于風(fēng)險(xiǎn)的現(xiàn)場(chǎng)核查�。因此,申辦者應(yīng)根據(jù)監(jiān)管法規(guī)動(dòng)態(tài)和現(xiàn)場(chǎng)核查要求�,及時(shí)調(diào)整合規(guī)管理體系�,嚴(yán)格依法開展臨床研究。
(二)外協(xié)管理
臨床研究涉及面廣�,申辦者一般需要外部服務(wù)機(jī)構(gòu)協(xié)助完成相關(guān)工作�。申辦者應(yīng)建立第三方服務(wù)機(jī)構(gòu)篩選�、合同管理等合規(guī)制度,選擇合格的第三方服務(wù)機(jī)構(gòu)并與其簽署權(quán)責(zé)明確的合同�,以監(jiān)督其履行職責(zé)。
(三)內(nèi)控制度
為規(guī)范臨床研究合規(guī)開展�,申辦者可在企業(yè)內(nèi)部建立相關(guān)的內(nèi)控制度,并要求各參與方在臨床研究開展過程中遵守相關(guān)的內(nèi)控要求�。
二、臨床參與方合規(guī)管理
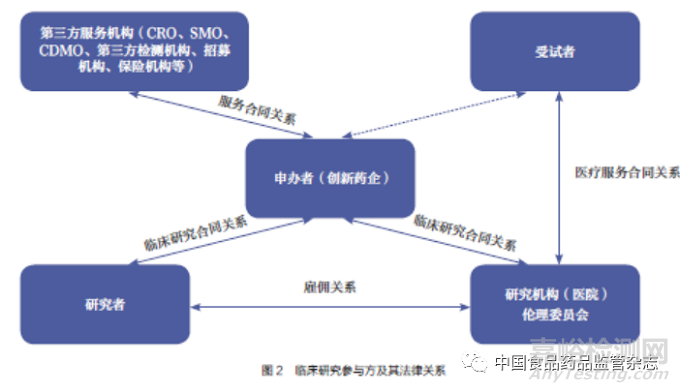
臨床研究涉及多方參與�,包括但不限于申辦者、醫(yī)療機(jī)構(gòu)/ 研究者�、受試者、合同研究組織(contract research organization�,CRO)、現(xiàn)場(chǎng)管理組織(site management organization�,SMO)�、第三方檢測(cè)機(jī)構(gòu)�、保險(xiǎn)公司、招募機(jī)構(gòu)等�。對(duì)于申辦者而言,與其他參與方簽署權(quán)責(zé)明確的合同并有效開展參與方合規(guī)管理�,是關(guān)乎臨床研究最終質(zhì)量的重要問題。臨床研究各參與方及其法律關(guān)系如圖2 所示�。
(一)研究機(jī)構(gòu)/ 研究者合規(guī)管理
申辦者如何選擇合適的研究機(jī)構(gòu)和研究者、避免利益沖突�、簽署權(quán)責(zé)明確的臨床研究合同、有效管理研究機(jī)構(gòu)和研究者履行職責(zé)�,是開展臨床研究需要重點(diǎn)關(guān)注的合規(guī)問題。
1. 資質(zhì)和內(nèi)控制度核查
根據(jù)GCP 和相關(guān)法律規(guī)定�,研究機(jī)構(gòu)和研究者應(yīng)具備并維持與開展臨床研究有關(guān)的資質(zhì)要求,并完成備案�。申辦者在臨床研究開始前可核查研究機(jī)構(gòu)和研究者是否具備相關(guān)資質(zhì),并在“藥物臨床試驗(yàn)機(jī)構(gòu)備案管理信息平臺(tái)” [2] 確認(rèn)研究機(jī)構(gòu)�、擬開展臨床研究所在科室和專業(yè)及主要研究者(principal investigator,PI)是否已完成備案�。
根據(jù)GCP 要求,臨床研究的實(shí)施應(yīng)當(dāng)遵守利益沖突回避原則�;根據(jù)《公立醫(yī)院內(nèi)部控制管理辦法》相關(guān)規(guī)定[3],公立醫(yī)療機(jī)構(gòu)應(yīng)當(dāng)建立臨床研究相關(guān)內(nèi)部控制制度�。申辦者可在臨床研究開始前要求研究機(jī)構(gòu)提供相關(guān)內(nèi)控制度并核查其是否履行�,包括但不限于臨床研究利益沖突政策�、臨床研究運(yùn)行管理制度�、臨床研究立項(xiàng)審批和審查制度、臨床研究經(jīng)費(fèi)使用管理制度以及臨床研究檔案管理制度等�,以判斷研究機(jī)構(gòu)是否在內(nèi)控層面滿足相關(guān)法律法規(guī)和臨床研究實(shí)施要求。
2. 合同管理
申辦者需要與研究機(jī)構(gòu)及主要研究者簽署權(quán)責(zé)明確的合同�,可在合同中重點(diǎn)關(guān)注以下內(nèi)容:①主要研究者的簽字確認(rèn)。②各方應(yīng)當(dāng)避免的�、可能的利益沖突要求。③研究者的職責(zé)�,如熟知研究方案、做好知情同意�、及時(shí)完善病例報(bào)告表等。④發(fā)生不良事件時(shí)研究機(jī)構(gòu)/ 研究者的救助和報(bào)告義務(wù)�、受試者權(quán)益保護(hù)內(nèi)容。⑤數(shù)據(jù)記錄和報(bào)告程序�。⑥申辦者的監(jiān)查和稽查、藥品監(jiān)督管理部門的檢查可直接去到試驗(yàn)現(xiàn)場(chǎng)�,查閱源數(shù)據(jù)、源文件和報(bào)告�。⑦研究經(jīng)費(fèi)合理且符合市場(chǎng)規(guī)律,建議明確每例受試者相關(guān)治療費(fèi)�、檢查費(fèi)、補(bǔ)償費(fèi)等費(fèi)用標(biāo)準(zhǔn)�,并合理設(shè)計(jì)支付節(jié)點(diǎn);對(duì)于篩選失敗�、失訪和脫落病例也建議約定結(jié)算方法�。⑧必備文件保存時(shí)間�、費(fèi)用和到期后的處理方法。⑨研究用藥物的儲(chǔ)存�、分發(fā)、使用�、返還等規(guī)定。⑩受試者個(gè)人信息保護(hù)措施和要求�。?生物樣本檢測(cè)要求。?臨床數(shù)據(jù)所有權(quán)及其他研究成果(包括探索性研究)的知識(shí)產(chǎn)權(quán)歸屬和對(duì)外發(fā)表文章的要求�。?各方的違約責(zé)任及免責(zé)情形。?合同終止情形�、終止程序和工作交接、對(duì)受試者的處理�、報(bào)告要求。?反腐敗反商業(yè)賄賂要求等�。
申辦者需要在合同中平衡各方利益,為臨床研究的合規(guī)開展預(yù)設(shè)相關(guān)的要求和標(biāo)準(zhǔn)�,并對(duì)合同履行實(shí)施監(jiān)督。
3. 監(jiān)查和稽查
監(jiān)查是申辦者對(duì)臨床研究進(jìn)行質(zhì)量控制的方法之一�。在GCP要求的常規(guī)監(jiān)查內(nèi)容外,為確保研究機(jī)構(gòu)/ 研究者合規(guī)開展臨床研究�,筆者認(rèn)為申辦者在監(jiān)查過程中還可額外關(guān)注以下內(nèi)容:①是否遵守研究方案和合同約定開展臨床研究。②研究者文件夾文件資料是否完整�、實(shí)時(shí)更新、準(zhǔn)確。③知情同意過程是否有記錄�,是否按照研究方案及相關(guān)法規(guī)要求進(jìn)行。④知情同意書和受試者鑒認(rèn)代碼表簽署時(shí)間是否存在邏輯關(guān)系�、簽字字跡是否一致�。⑤登錄醫(yī)院信息系統(tǒng),核查是否按規(guī)定要求進(jìn)行受試者訪視�。⑥安全性數(shù)據(jù)及記錄,確認(rèn)有無嚴(yán)重不良事件發(fā)生�。⑦記錄前后的一致性,有無矛盾或遺漏�,記錄是否準(zhǔn)確、充分�。⑧實(shí)驗(yàn)室檢查結(jié)果,尤其是異常結(jié)果的記錄和反饋情況�。⑨研究用藥物的相關(guān)使用記錄情況等。
除監(jiān)查外�,申辦者還可選定獨(dú)立于臨床研究的人員擔(dān)任稽查員,依據(jù)稽查計(jì)劃和規(guī)程對(duì)臨床研究相關(guān)活動(dòng)和文件進(jìn)行稽查�。
通過監(jiān)查和稽查,申辦者可以掌握研究機(jī)構(gòu)/ 研究者實(shí)施臨床研究的具體情況�,發(fā)現(xiàn)任何問題時(shí)可要求研究機(jī)構(gòu)/ 研究者及時(shí)進(jìn)行整改,防止影響臨床研究質(zhì)量�。
4. 境外研究機(jī)構(gòu)/ 研究者合規(guī)管理
在國(guó)際多中心臨床研究(multi-regional clinical trial,MRCT)中�,申辦者還需要關(guān)注不同國(guó)家和地區(qū)對(duì)于研究機(jī)構(gòu)/研究者的合規(guī)要求�。以美國(guó)為例�,美國(guó)食品藥品監(jiān)督管理局(Food and Drug Administration�,F(xiàn)DA) 要求申辦者應(yīng)公開臨床研究信息,實(shí)施公眾監(jiān)督�,即符合要求的藥物臨床研究必須在美國(guó)國(guó)立衛(wèi)生研究院建立的網(wǎng)站[4]上進(jìn)行注冊(cè),填報(bào)規(guī)定的基本信息�,否則申辦者將受到重罰。對(duì)于研究機(jī)構(gòu)/ 研究者�,F(xiàn)DA 要求其應(yīng)具備相關(guān)資質(zhì),研究者需要接受過臨床研究相關(guān)培訓(xùn)�,嚴(yán)格按照美國(guó)GCP 要求開展研究[5]。FDA 還會(huì)不定期發(fā)布針對(duì)研究者的警告信�,告誡研究者在臨床研究中的不合規(guī)之處,也使相關(guān)整改更具有針對(duì)性�。
根據(jù)近年來FDA 針對(duì)研究者的警告信內(nèi)容,F(xiàn)DA 對(duì)于研究者的監(jiān)管要求主要包括[6] :①能否按照計(jì)劃進(jìn)行研究�。②能否全面準(zhǔn)確記錄數(shù)據(jù)�。③能否獲取/ 記錄受試者同意。④能否妥善落實(shí)藥品管理�。⑤能否將更改通知倫理委員會(huì)�、提交進(jìn)度報(bào)告�、監(jiān)督或親自進(jìn)行臨床研究。⑥能否妥善保護(hù)受試者權(quán)益�。⑦是否獲得或記錄倫理委員會(huì)的批準(zhǔn)�。⑧能否報(bào)告藥物不良反應(yīng)�。⑨能否報(bào)告臨床研究結(jié)果等。
我國(guó)申辦者在涉及美國(guó)的多中心臨床研究中�����,可重點(diǎn)關(guān)注研究者能否履行上述合規(guī)要求�����,以保證臨床研究質(zhì)量�����。
(二)臨床服務(wù)機(jī)構(gòu)合規(guī)管理
根據(jù)GCP 第三十三條規(guī)定�����,申辦者可以將其臨床研究的部分或者全部工作和任務(wù)委托給合同研究組織�����,監(jiān)督合同研究組織承擔(dān)的各項(xiàng)工作�����。實(shí)踐中�����,除合同研究組織外�����, 申辦者還可能委托現(xiàn)場(chǎng)管理組織委派臨床研究協(xié)調(diào)員(clinical research coordinator�����,CRC) 為研究者提供相關(guān)服務(wù)�����、委托招募機(jī)構(gòu)進(jìn)行受試者招募�����、委托第三方檢測(cè)機(jī)構(gòu)對(duì)生物樣本進(jìn)行檢測(cè)等�����。涉及國(guó)際多中心臨床研究時(shí)�����,申辦者也可能委托境外的服務(wù)機(jī)構(gòu)履行相關(guān)工作�����。
申辦者如何選擇合適的第三方服務(wù)機(jī)構(gòu)�����、與其簽署權(quán)責(zé)明確的合同�����、監(jiān)督和管理其履行職責(zé)是必須要解決的合規(guī)難題�����。本文從合同研究組織�����、現(xiàn)場(chǎng)管理組織�����、招募機(jī)構(gòu)�����、檢測(cè)機(jī)構(gòu)為切入點(diǎn)�����,探討上述合規(guī)問題的解決路徑�����。
1. 合同研究組織
(1)盡職調(diào)查�����。申辦者在選聘合同研究組織之前可以對(duì)其進(jìn)行盡職調(diào)查�����,了解其基本信息�����、相關(guān)業(yè)務(wù)資質(zhì)、服務(wù)報(bào)價(jià)�����、既往服務(wù)案例�����、內(nèi)控制度及執(zhí)行情況(如反腐敗反商業(yè)賄賂制度�����、臨床研究項(xiàng)目管理制度�����、商業(yè)秘密和知識(shí)產(chǎn)權(quán)保護(hù)制度等)�����,綜合對(duì)比后選擇合適的合同研究組織�����。申辦者還可以建立合同研究組織的篩選制度�����,明確相關(guān)的招投標(biāo)機(jī)制�����,防止因選聘合同研究組織過程中的商業(yè)賄賂問題最終影響臨床研究質(zhì)量�����。
(2) 合同管理�����。合同研究組織通過與申辦者簽署的服務(wù)合同參與到臨床研究中�����,若合同條款約定不明�����、權(quán)責(zé)劃分不清�����,很可能在實(shí)踐中產(chǎn)生糾紛,影響臨床研究進(jìn)度�����。因此�����,申辦者需要在合同中明確雙方的權(quán)責(zé)�����,并可重點(diǎn)關(guān)注以下幾點(diǎn)內(nèi)容:①申辦者委托的具體工作以及相應(yīng)的標(biāo)準(zhǔn)操作規(guī)程�����。②項(xiàng)目人員和資源配備情況�����、人員更換流程�����、雙方溝通層級(jí)和頻率�����。③合理設(shè)計(jì)費(fèi)用支付節(jié)點(diǎn)�����、支付要求和支付比例�����,明確代墊費(fèi)用范疇及支付流程�����,明確備用金金額�����、使用流程和結(jié)算制度(如需)�����。④明確受試者個(gè)人信息保護(hù)和數(shù)據(jù)安全要求。⑤明確記錄�����、報(bào)告和監(jiān)管協(xié)助要求�����。⑥明確知識(shí)產(chǎn)權(quán)的歸屬情況�����。⑦明確合同終止情形�����、單方終止權(quán)利的安排�����、終止后受試者安排和工作移交程序等�����。⑧明確違約責(zé)任承擔(dān)主體和范圍及免責(zé)事項(xiàng)�����;明確對(duì)第三方供應(yīng)商的監(jiān)督或管理責(zé)任及相關(guān)的違約責(zé)任安排�����。⑨明確質(zhì)量控制和保證具體要求�����,明確申辦者的監(jiān)查和稽查以及審計(jì)權(quán)利�����。⑩可以要求合同研究組織簽署廉潔承諾函�����,爭(zhēng)取合規(guī)風(fēng)險(xiǎn)隔離等�����。
在國(guó)際多中心臨床研究中�����,申辦者可將臨床研究相關(guān)工作委托給業(yè)務(wù)領(lǐng)域能夠覆蓋多中心的合同研究組織,并與其簽署權(quán)責(zé)明確的合同�����。在此類合同中�����,申辦者除需要關(guān)注以上所列內(nèi)容外�����,還需要關(guān)注不同地區(qū)對(duì)于臨床研究監(jiān)管的特殊要求并注意適用法律的選擇�����。
2. 現(xiàn)場(chǎng)管理組織
在我國(guó)臨床研究中�����,申辦者通常會(huì)聘請(qǐng)第三方獨(dú)立的現(xiàn)場(chǎng)管理組織�����,并由其派遣合格的臨床研究協(xié)調(diào)員協(xié)助研究者進(jìn)行臨床研究中非醫(yī)學(xué)判斷的事務(wù)性和規(guī)范性工作�����。在選聘現(xiàn)場(chǎng)管理組織之前,申辦者應(yīng)充分考慮研究機(jī)構(gòu)和研究者的意見�����。申辦者可與研究機(jī)構(gòu)�����、現(xiàn)場(chǎng)管理組織簽署三方合同�����,明確各方的權(quán)利義務(wù)�����;在得到研究機(jī)構(gòu)和研究者同意的前提下�����,申辦者也可直接與現(xiàn)場(chǎng)管理組織簽署相關(guān)合同�����。申辦者在與現(xiàn)場(chǎng)管理組織簽署的合同中可以明確以下內(nèi)容:①現(xiàn)場(chǎng)管理組織及其委派的臨床研究協(xié)調(diào)員資質(zhì)要求�����。②臨床研究協(xié)調(diào)員的委派和更換程序�����。③臨床研究協(xié)調(diào)員的工作內(nèi)容和匯報(bào)要求�����,要特別注意其不得從事涉及醫(yī)學(xué)判斷的事務(wù)和工作�����。④現(xiàn)場(chǎng)管理組織與臨床研究協(xié)調(diào)員的雇傭關(guān)系�����。⑤患者健康信息�����、醫(yī)療信息和其他個(gè)人信息保護(hù)要求�����。⑥付款計(jì)劃與受試者入組和訪視進(jìn)度相協(xié)調(diào)。⑦ 保密和知識(shí)產(chǎn)權(quán)歸屬�����。⑧違約責(zé)任和救濟(jì)措施等�����。
成熟市場(chǎng)的現(xiàn)場(chǎng)管理組織服務(wù)方式更加多元化�����。例如美國(guó)的現(xiàn)場(chǎng)管理組織可以憑借其臨床信息資源網(wǎng)絡(luò)和專業(yè)經(jīng)驗(yàn)�����,加速研究機(jī)構(gòu)和研究者的選擇及資格評(píng)估�����、合同談判和研究啟動(dòng)等過程, 幫助申辦者提高效率�����。對(duì)于研究機(jī)構(gòu)而言, 現(xiàn)場(chǎng)管理組織可以為其拓展業(yè)務(wù)�����,幫助其進(jìn)入全球研究者網(wǎng)絡(luò)�����,使研究機(jī)構(gòu)有更多機(jī)會(huì)參與國(guó)際多中心臨床研究?����,F(xiàn)場(chǎng)管理組織還可以參與研究方案的可行性分析�����、遞交倫理委員會(huì)申請(qǐng)�����、實(shí)施受試者招募和知情同意�����、收集并記錄數(shù)據(jù)、參與臨床研究現(xiàn)場(chǎng)標(biāo)準(zhǔn)操作規(guī)程制定�����、參與研究者和研究人員培訓(xùn)工作等[7]�����。在開展國(guó)際多中心臨床研究時(shí)�����,申辦者也可以選擇境外專業(yè)現(xiàn)場(chǎng)管理組織�����, 提供更多元化服務(wù)�����,提高臨床研究效率�����。
3. 招募機(jī)構(gòu)
傳統(tǒng)受試者招募方式(如研究者現(xiàn)場(chǎng)招募�����、通過招募廣告等途徑公開招募)可能存在受試者招募數(shù)量不足�����、入組進(jìn)度緩慢等情況�����,由此催生了商業(yè)性招募機(jī)構(gòu)的發(fā)展�����。商業(yè)性招募機(jī)構(gòu)實(shí)際上提供的是信息中介服務(wù)�����,由招募機(jī)構(gòu)將潛在受試者信息推薦給研究者�����,最終根據(jù)推薦數(shù)量或推薦成功數(shù)量�����,由申辦者支付招募機(jī)構(gòu)相應(yīng)費(fèi)用。在這種招募模式中�����,如何確保招募信息來源的合規(guī)性�����、如何保證招募過程中受試者個(gè)人信息保護(hù)�����、如何防止招募機(jī)構(gòu)與研究者或受試者間的商業(yè)賄賂等問題�����,是商業(yè)性招募的合規(guī)痛點(diǎn)問題�����。在國(guó)際多中心臨床研究中�����,申辦者還面臨境外招募受試者的問題�����。本文基于申辦者視角�����,嘗試性地探討上述痛點(diǎn)問題的解決路徑�����。
(1)招募信息來源合規(guī)性問題�����。招募機(jī)構(gòu)在獲取潛在受試者個(gè)人信息過程中�����,要特別注意受試者個(gè)人隱私保護(hù)問題�����。招募機(jī)構(gòu)不應(yīng)在未獲得個(gè)人知情同意的前提下�����,直接接觸潛在受試者個(gè)人身份信息、醫(yī)療信息等隱私信息�����。在通過潛在受試者主治醫(yī)生獲得受試者個(gè)人信息前�����,不應(yīng)由主治醫(yī)生同意就直接獲取受試者個(gè)人信息�����,而應(yīng)由主治醫(yī)生詢問潛在受試者個(gè)人意愿�����,在潛在受試者愿意了解臨床研究相關(guān)信息且簽署知情同意書后�����,招募機(jī)構(gòu)才可以獲取潛在受試者個(gè)人信息�����;在通過檢測(cè)實(shí)驗(yàn)室或其他數(shù)據(jù)庫(kù)數(shù)據(jù)識(shí)別潛在受試者前�����,應(yīng)在獲得潛在受試者知情同意后才可將個(gè)人信息提供給招募機(jī)構(gòu)�����。招募機(jī)構(gòu)將潛在受試者的個(gè)人信息推薦給研究者前�����,均應(yīng)獲得潛在受試者知情同意�����。
(2)受試者隱私保護(hù)和反商業(yè)賄賂�����。申辦者在與招募機(jī)構(gòu)簽署的合同中可對(duì)受試者隱私保護(hù)和商業(yè)賄賂問題進(jìn)行約定�����,并保留申辦者對(duì)招募機(jī)構(gòu)履約行為進(jìn)行監(jiān)督和審計(jì)的權(quán)利�����。建議申辦者可在相關(guān)合同中明確以下幾點(diǎn)內(nèi)容:①招募機(jī)構(gòu)承諾遵守受試者隱私保護(hù)和反商業(yè)賄賂相關(guān)法規(guī)政策,并約定相關(guān)的違約責(zé)任�����。②招募機(jī)構(gòu)不得參與一切與受試者招募有關(guān)的醫(yī)學(xué)判斷�����。③招募機(jī)構(gòu)提供的服務(wù)不得超出經(jīng)倫理委員會(huì)批準(zhǔn)的研究方案及招募信息/ 廣告的范圍�����。④招募機(jī)構(gòu)不得未經(jīng)個(gè)人知情同意即接觸和披露個(gè)人隱私信息�����。⑤招募機(jī)構(gòu)不得實(shí)施任何行為影響潛在受試者意愿或使受試者退出�����。⑥招募機(jī)構(gòu)不得向任何受試者/ 潛在受試者收取任何費(fèi)用或回報(bào)等�����。⑦申辦者對(duì)招募機(jī)構(gòu)的監(jiān)督和審計(jì)權(quán)利等�����。
在國(guó)際多中心臨床研究中�����,需要招募不同國(guó)家和地區(qū)的受試者�����,因此申辦者也需要了解其他國(guó)家和地區(qū)臨床研究中受試者招募方式�����。以美國(guó)為例�����,其在受試者招募中一般采取多種招募方式以加快招募速度�����,如在各類媒體上發(fā)布各種類型的招募廣告�����、開展針對(duì)公眾的招募宣傳活動(dòng)、利用研究機(jī)構(gòu)及現(xiàn)場(chǎng)管理組織進(jìn)行招募�����、通過患者組織(patient organizations)進(jìn)行招募等�����。此外�����,F(xiàn)DA 在國(guó)際多中心臨床研究中越來越關(guān)注對(duì)于引起地區(qū)差異等潛在因素的研究和分析的重要性[8]�����。因此�����,對(duì)于開展國(guó)際多中心臨床研究的申辦者而言�����,需要特別關(guān)注不同區(qū)域和人種的受試者招募,在符合各個(gè)地區(qū)相關(guān)法律法規(guī)的前提下通過多種方式招募受試者�����。
4. 檢測(cè)機(jī)構(gòu)
(1)盡職調(diào)查�����。臨床研究中若涉及生物標(biāo)志物�����、基因測(cè)序�����、藥物代謝動(dòng)力學(xué)�����、藥效動(dòng)力學(xué)等檢測(cè)項(xiàng)目�����,申辦者可能會(huì)委托第三方檢測(cè)機(jī)構(gòu)來進(jìn)行檢測(cè)�����。申辦者在選聘檢測(cè)機(jī)構(gòu)之前�����,可對(duì)檢測(cè)機(jī)構(gòu)進(jìn)行盡職調(diào)查�����,了解檢測(cè)機(jī)構(gòu)的基本信息�����、既往服務(wù)案例�����、內(nèi)部的質(zhì)量保證和控制制度�����、隱私保護(hù)政策及相關(guān)的運(yùn)行情況�����,以及是否取得相關(guān)資質(zhì)[ 如中國(guó)合格評(píng)定國(guó)家認(rèn)可委員會(huì)(ChinaNational Accreditation Service for Conformity Assessment,CNAS) 關(guān)于ISO 15189 質(zhì)量管理體系的認(rèn)可�����、美國(guó)病理學(xué)家協(xié)會(huì)(College of AmericanPathologists�����,CAP)�����、《美國(guó)臨床實(shí)驗(yàn)室改進(jìn)法案修正案》(clinical laboratory improvement amendments�����,CLIA)的認(rèn)證等]�����。申辦者還應(yīng)當(dāng)在知情同意書中明確告知受試者采集的樣本類型�����、檢測(cè)項(xiàng)目和用途�����、第三方檢測(cè)機(jī)構(gòu)等信息�����,并就受試者的生物樣本第三方檢測(cè)獲得受試者知情同意�����。
(2)合同管理�����。申辦者應(yīng)與檢測(cè)機(jī)構(gòu)簽署權(quán)責(zé)明確的合同�����,并可在合同中明確以下內(nèi)容:①樣本的采集和運(yùn)輸要求以及樣本損毀的責(zé)任承擔(dān)�����。②樣本類型�����、例數(shù)、檢測(cè)技術(shù)�����、檢測(cè)結(jié)果交付�����、剩余樣本處理�����、數(shù)據(jù)保存等要求�����。③檢測(cè)費(fèi)用組成�����、支付節(jié)點(diǎn)及付款條件�����。④檢測(cè)機(jī)構(gòu)配合藥品監(jiān)管部門檢查的義務(wù)�����。⑤檢測(cè)工作成果的知識(shí)產(chǎn)權(quán)歸屬等�����。
在國(guó)際多中心臨床研究中�����,若涉及質(zhì)控樣本的交叉驗(yàn)證�����,申辦者可選擇業(yè)務(wù)能夠覆蓋國(guó)際多中心的檢測(cè)機(jī)構(gòu)�����,以使交叉驗(yàn)證能夠順利進(jìn)行并保證檢測(cè)方法的一致性�����。若涉及人類遺傳資源材料出入境和海關(guān)進(jìn)出口�����,申辦者還應(yīng)根據(jù)人類遺傳資源管理的相關(guān)法規(guī)完成相關(guān)審批或備案程序、按照海關(guān)管理的相關(guān)法規(guī)完成海關(guān)進(jìn)出口手續(xù)等�����。
三�����、申辦者內(nèi)控制度建設(shè)
除針對(duì)臨床參與方的合規(guī)管理外�����,申辦者還需要建立完善的內(nèi)部臨床研究合規(guī)管理制度體系�����,以覆蓋臨床研究的全生命周期�����。這些制度包括但不限于數(shù)據(jù)合規(guī)管理制度�����、人類遺傳資源管理制度�����、第三方服務(wù)機(jī)構(gòu)選聘制度�����、質(zhì)量管理與質(zhì)量保證制度等�����。申辦者可基于管理制度制定相關(guān)的標(biāo)準(zhǔn)操作規(guī)程�����,如試驗(yàn)數(shù)據(jù)記錄�����、受試者知情同意�����、不良事件及嚴(yán)重不良事件報(bào)告和處理�����、實(shí)驗(yàn)室檢測(cè)及質(zhì)量控制等標(biāo)準(zhǔn)操作規(guī)程。本文通過節(jié)選臨床數(shù)據(jù)合規(guī)和人類遺傳資源管理的相關(guān)內(nèi)容�����,探討申辦者應(yīng)如何建立相關(guān)的合規(guī)管理制度�����。
(一)臨床研究數(shù)據(jù)合規(guī)管理
臨床研究數(shù)據(jù)是評(píng)估藥物安全性�����、有效性的關(guān)鍵考量因素�����。如何降低數(shù)據(jù)合規(guī)風(fēng)險(xiǎn)并提高數(shù)據(jù)質(zhì)量�����、如何解決國(guó)際多中心臨床研究數(shù)據(jù)分享等數(shù)據(jù)合規(guī)問題�����,是申辦者亟需解決的問題�����。
1. 數(shù)據(jù)合規(guī)監(jiān)管體系
臨床研究數(shù)據(jù)合規(guī)監(jiān)管體系以《藥品管理法》《生物安全法》《網(wǎng)絡(luò)安全法》《數(shù)據(jù)安全法》《個(gè)人信息保護(hù)法》等一般藥品管理�����、生物安全�����、網(wǎng)絡(luò)安全�����、數(shù)據(jù)安全�����、個(gè)人信息保護(hù)法律為基礎(chǔ)�����,以GCP、《人類遺傳資源管理?xiàng)l例》《人口健康信息管理辦法(試行)》《國(guó)家健康醫(yī)療大數(shù)據(jù)標(biāo)準(zhǔn)�����、安全和服務(wù)管理辦法(試行)》和GB/T 39725-2020《信息安全技術(shù) 健康醫(yī)療數(shù)據(jù)安全指南》等醫(yī)藥醫(yī)療與生命科學(xué)領(lǐng)域數(shù)據(jù)管理的單行法規(guī)�����、條例�����、國(guó)家標(biāo)準(zhǔn)和指南為具體規(guī)則�����,涉及國(guó)家市場(chǎng)監(jiān)督管理總局�����、科學(xué)技術(shù)部�����、工業(yè)和信息化部等監(jiān)管部門�����。申辦者應(yīng)在了解數(shù)據(jù)合規(guī)監(jiān)管體系的基礎(chǔ)上,進(jìn)一步完善數(shù)據(jù)合規(guī)管理制度�����。
2. 數(shù)據(jù)合規(guī)常見問題
申辦者是藥品注冊(cè)的申請(qǐng)者和權(quán)利人�����,對(duì)臨床研究數(shù)據(jù)可靠性承擔(dān)法律責(zé)任�����。藥品監(jiān)管機(jī)構(gòu)開展的臨床研究數(shù)據(jù)核查工作反映了諸多數(shù)據(jù)合規(guī)問題�����,申辦者可以在了解相關(guān)的合規(guī)問題后“有的放矢”�����,建立有效的合規(guī)管理體系�����。常見的數(shù)據(jù)合規(guī)問題主要包括:①編造或者無合理解釋地修改受試者信息以及研究數(shù)據(jù)�����、研究記錄�����、研究藥物信息�����。②隱瞞研究數(shù)據(jù)�����,無合理解釋地棄用研究數(shù)據(jù)�����,以其他方式違反研究方案選擇性使用的研究數(shù)據(jù)�����。③瞞報(bào)與臨床研究用藥相關(guān)或可能相關(guān)的嚴(yán)重不良事件�����。④故意損毀、隱匿臨床研究數(shù)據(jù)或者數(shù)據(jù)存儲(chǔ)介質(zhì)�����。⑤受試者的篩選/ 入組相關(guān)數(shù)據(jù)鏈缺乏完整性�����。⑥知情同意書的簽署內(nèi)容不完整�����、不規(guī)范�����,簽署數(shù)量與總結(jié)報(bào)告中的篩選和入選病例數(shù)不一致�����。⑦臨床研究過程記錄及臨床檢查�����、化驗(yàn)等數(shù)據(jù)不真實(shí)且無法溯源�����。⑧電子數(shù)據(jù)的修改�����、轉(zhuǎn)換�����、編輯未留痕且不可追溯�����。⑨臨床研究的生物樣本采集�����、保存�����、運(yùn)送與交接記錄無法保證完整性和真實(shí)性�����。⑩研究用藥物的管理過程記錄缺乏完整性和原始性等。
3. 國(guó)際多中心臨床研究數(shù)據(jù)合規(guī)
國(guó)際多中心臨床研究的數(shù)據(jù)合規(guī)主要涉及數(shù)據(jù)出境及域外認(rèn)可問題�����。
對(duì)于數(shù)據(jù)出境�����,需要在數(shù)據(jù)合規(guī)監(jiān)管體系下�����,根據(jù)數(shù)據(jù)的不同屬性適用不同的監(jiān)管法規(guī)要求�����。如涉及重要數(shù)據(jù)�����、10 萬人個(gè)人信息或者1 萬人敏感個(gè)人信息等情況�����,需要按照相關(guān)法律法規(guī)的要求進(jìn)行數(shù)據(jù)出境安全評(píng)估�����;涉及人類遺傳資源的數(shù)據(jù)�����,應(yīng)履行相關(guān)的信息備份和備案程序�����;如涉及人口健康信息的�����,跨境傳輸將被禁止�����。
對(duì)于數(shù)據(jù)域外認(rèn)可�����, 根據(jù)《接受藥品境外臨床試驗(yàn)數(shù)據(jù)的技術(shù)指導(dǎo)原則》的要求�����, 若境外臨床研究數(shù)據(jù)真實(shí)可靠、符合ICH- GCP 和藥品注冊(cè)檢查要求�����、支持目標(biāo)適應(yīng)癥的有效性和安全性評(píng)價(jià)�����、不存在影響有效性和安全性的種族敏感性因素等�����,則可被國(guó)家藥品監(jiān)管部門完全接受�����。根據(jù)美國(guó)2012 年《FDA安全和創(chuàng)新法案》(Food and Drug Administration Safety and Innovation Act�����,F(xiàn)DASIA) 的規(guī)定以及FDA 網(wǎng)站公布的接受境外藥品臨床研究數(shù)據(jù)的指引�����,若數(shù)據(jù)能夠被FDA 驗(yàn)證�����、具有有能力的研究者�����、根據(jù)美國(guó)GCP 的規(guī)定進(jìn)行且適用于美國(guó)公民及醫(yī)療實(shí)踐等�����,則可被FDA 接受[9]�����。
近期�����,美國(guó)曾質(zhì)詢過我國(guó)制藥企業(yè)使用在我國(guó)進(jìn)行的/ 主要在我國(guó)進(jìn)行的臨床研究數(shù)據(jù)來提交新藥申請(qǐng)�����,其從側(cè)面反映我國(guó)制藥企業(yè)走出國(guó)門仍面臨諸多困難。對(duì)于要成為全球化的制藥企業(yè)而言�����,更需要密切關(guān)注不同區(qū)域臨床數(shù)據(jù)合規(guī)要求和域外認(rèn)可標(biāo)準(zhǔn)�����,建立并完善數(shù)據(jù)合規(guī)管理制度�����,合規(guī)開展國(guó)際多中心臨床研究�����。
4. 數(shù)據(jù)合規(guī)管理制度建設(shè)
為了使臨床研究數(shù)據(jù)合法合規(guī)收集�����、使用�����,申辦者可以建立相關(guān)的數(shù)據(jù)合規(guī)管理制度�����,重點(diǎn)可關(guān)注以下內(nèi)容:①建立數(shù)據(jù)管理相關(guān)人員的責(zé)任�����、資質(zhì)及培訓(xùn)制度�����,明確申辦者�����、研究者�����、監(jiān)查員�����、數(shù)據(jù)管理員�����、合同研究組織的責(zé)任,并進(jìn)行定期評(píng)估及考核�����。②建立臨床研究數(shù)據(jù)管理系統(tǒng)�����,包括質(zhì)量手冊(cè)�����、程序文件�����、作業(yè)指導(dǎo)書�����、質(zhì)量記錄等�����,并確保數(shù)據(jù)的可溯源性�����。③建立數(shù)據(jù)標(biāo)準(zhǔn)化體系�����,為申辦者之間的交流�����、申辦者與藥物評(píng)審機(jī)構(gòu)之間的交流及各臨床研究的藥物安全性數(shù)據(jù)共享提供便利�����。④建立數(shù)據(jù)質(zhì)量的保障及評(píng)估制度�����,包括數(shù)據(jù)核查制度�����、數(shù)據(jù)質(zhì)疑流程�����、數(shù)據(jù)修改流程、數(shù)據(jù)盲態(tài)審核等�����。⑤建立安全性數(shù)據(jù)及嚴(yán)重不良事件報(bào)告制度�����,包括為確保嚴(yán)重不良事件數(shù)據(jù)的一致性而對(duì)臨床研究數(shù)據(jù)庫(kù)與藥物警戒數(shù)據(jù)庫(kù)的一致性核查等�����。
除以上數(shù)據(jù)管理制度外�����,臨床研究中所收集�����、存儲(chǔ)�����、使用的數(shù)據(jù)信息還可能涉及個(gè)人隱私信息�����、健康醫(yī)療大數(shù)據(jù)、人類遺傳資源信息或人口健康信息�����。這些數(shù)據(jù)的存儲(chǔ)和傳輸都有特定的法規(guī)要求�����。申辦者需要結(jié)合具體實(shí)踐情況�����,綜合考慮數(shù)據(jù)合規(guī)監(jiān)管體系的各項(xiàng)具體要求�����,合法合規(guī)地收集�����、存儲(chǔ)和使用臨床研究中的各項(xiàng)數(shù)據(jù)�����。
(二)人類遺傳資源合規(guī)管理
臨床研究通常需要采集人體血液�����、腦脊液�����、細(xì)胞等生物樣本�����,可能涉及人類遺傳資源�����,需要履行人類遺傳資源相關(guān)的審批或備案程序�����。實(shí)踐中�����,如何認(rèn)定“外方單位”�����,如何合規(guī)開展國(guó)際合作科學(xué)研究,以及如何在企業(yè)內(nèi)部建立人類遺傳資源合規(guī)管理制度�����,是申辦者面臨的合規(guī)難題�����。
1. 外方單位認(rèn)定
《人類遺傳資源管理?xiàng)l例》(以下簡(jiǎn)稱《條例》)中明確規(guī)定�����,外方單位是指外國(guó)組織及外國(guó)組織�����、個(gè)人設(shè)立或者實(shí)際控制的機(jī)構(gòu)�����?����!度祟愡z傳資源管理?xiàng)l例實(shí)施細(xì)則(征求意見稿)》(以下簡(jiǎn)稱《實(shí)施細(xì)則》)第十二條對(duì)于“實(shí)際控制”一詞限定為四種情形�����,對(duì)于外方單位的認(rèn)定更偏向于從實(shí)質(zhì)層面上認(rèn)定境外組織�����、個(gè)人是否足以對(duì)機(jī)構(gòu)的決策�����、管理等重大事項(xiàng)施加重大影響�����,而不是單純從形式層面上以股權(quán)占比作為認(rèn)定標(biāo)準(zhǔn)�����。但《實(shí)施細(xì)則》對(duì)于“設(shè)立”尚未進(jìn)一步細(xì)化解釋�����。
關(guān)于外方單位的認(rèn)定,在目前的實(shí)際操作中�����,任何向上追溯含有外資成分的企業(yè)都可能會(huì)被中國(guó)人類遺傳資源管理辦公室(以下簡(jiǎn)稱遺傳辦)認(rèn)定為外方單位�����,且通過協(xié)議控制方式被控制的境內(nèi)運(yùn)營(yíng)實(shí)體也會(huì)被認(rèn)定為外方單位�����。但是基于《實(shí)施細(xì)則》中對(duì)“實(shí)際控制”認(rèn)定標(biāo)準(zhǔn)作出的進(jìn)一步限定�����,未來實(shí)操中認(rèn)定外方單位的標(biāo)準(zhǔn)可能會(huì)發(fā)生變化�����,申辦者需要密切關(guān)注《實(shí)施細(xì)則》的更新發(fā)布情況�����,以進(jìn)一步判斷企業(yè)在人類遺傳資源監(jiān)管體系下的性質(zhì)�����。
2. 國(guó)際合作科學(xué)研究合規(guī)管理
國(guó)際合作科學(xué)研究是目前含有外資成分的申辦者利用我國(guó)人類遺傳資源開展臨床研究的可行途徑�����,需要申辦者了解國(guó)際合作科學(xué)研究的審批/ 備案要求�����、國(guó)際合作科學(xué)研究的知識(shí)產(chǎn)權(quán)歸屬及人類遺傳資源信息對(duì)外提供等問題�����。
(1)審批/ 備案要求�����。利用我國(guó)人類遺傳資源開展國(guó)際合作科學(xué)研究的�����,應(yīng)當(dāng)由中方單位和外方單位共同提出申請(qǐng)并經(jīng)遺傳辦批準(zhǔn)�����。為獲得相關(guān)藥品在我國(guó)上市許可,在臨床機(jī)構(gòu)利用我國(guó)人類遺傳資源開展國(guó)際合作科學(xué)研究�����、不涉及人類遺傳資源材料出境的�����, 不需要審批�����。但是�����,合作雙方在開展臨床試驗(yàn)前應(yīng)當(dāng)將擬使用的人類遺傳資源種類�����、數(shù)量及其用途向遺傳辦備案�����。對(duì)于研究者發(fā)起的臨床研究(investigator initiated trial�����,IIT)而言�����,若涉及利用人類遺傳資源的�����,也需要履行相關(guān)審批和備案程序�����。但實(shí)踐中外方單位開展的IIT 在遺傳辦審批中可能會(huì)遇到較大阻礙�����。
(2)知識(shí)產(chǎn)權(quán)歸屬�����?����!稐l例》規(guī)定,利用我國(guó)人類遺傳資源開展國(guó)際合作科學(xué)研究�����,產(chǎn)生的成果申請(qǐng)專利的�����,應(yīng)當(dāng)由中方單位和外方單位共同提出申請(qǐng)�����,專利權(quán)歸合作雙方共有�����。實(shí)踐中�����,遺傳辦特別關(guān)注探索性研究的知識(shí)產(chǎn)權(quán)共有問題�����。若申辦者為外方單位�����,其與中方單位簽署的合同中應(yīng)明確探索性研究的知識(shí)產(chǎn)權(quán)由合作雙方共享�����。
(3)信息對(duì)外提供�����?����!稐l例》規(guī)定�����,將人類遺傳資源信息向外方單位提供或者開放使用的�����,應(yīng)當(dāng)向遺傳辦備案并提交信息備份�����。根據(jù)《人類遺傳資源管理常見問題解答(系列問答二)》中的官方答復(fù),利用我國(guó)人類遺傳資源開展國(guó)際合作科學(xué)研究所產(chǎn)生的人類遺傳資源信息�����,合作雙方可以使用�����,不需進(jìn)行數(shù)據(jù)信息對(duì)外提供備案�����。參加合作的其他外方單位如要使用相關(guān)研究數(shù)據(jù)�����,則應(yīng)由合作雙方中的中方數(shù)據(jù)信息所有者申請(qǐng)數(shù)據(jù)信息對(duì)外提供或開放使用備案�����。另外�����,申辦者需注意,若針對(duì)臨床研究成果發(fā)表文章涉及開放使用人類遺傳資源信息的�����,應(yīng)在信息出境前進(jìn)行信息備份和備案�����。
3. 人類遺傳資源合規(guī)管理制度建設(shè)
由于人類遺傳資源監(jiān)管要求的復(fù)雜性�����,常會(huì)影響臨床研究的整體進(jìn)度�����,因此申辦者有必要在內(nèi)部建立人類遺傳資源合規(guī)管理制度�����,以高效管理人類遺傳資源�����,推進(jìn)臨床研究項(xiàng)目進(jìn)度�����。申辦者可根據(jù)企業(yè)性質(zhì)�����,按照以下“三步法”來搭建人類遺傳資源合規(guī)管理制度�����。
第一�����,業(yè)務(wù)梳理與評(píng)估�����。首先判斷企業(yè)以及業(yè)務(wù)合作方的主體性質(zhì)�����,是中方單位還是外方單位�����;其次,梳理目前開展的所有業(yè)務(wù)類型�����,明確哪些業(yè)務(wù)涉及人類遺傳資源和涉及的環(huán)節(jié)(如人類遺傳資源采集�����、保藏�����、利用等)�����;最后�����,對(duì)業(yè)務(wù)進(jìn)行風(fēng)險(xiǎn)等級(jí)評(píng)估�����,按照人類遺傳資源不同的監(jiān)管要求進(jìn)行高�����、中�����、低風(fēng)險(xiǎn)劃分�����。
第二�����,內(nèi)部管理體系搭建�����。根據(jù)企業(yè)管理架構(gòu)�����,明確負(fù)責(zé)人類遺傳資源管理工作的部門及承擔(dān)的職責(zé)�����,并根據(jù)不同的業(yè)務(wù)風(fēng)險(xiǎn)等級(jí)劃分,確定相應(yīng)的審批部門并制定相應(yīng)的審批流程�����。
第三�����,體系化管理制度建設(shè)�����。根據(jù)業(yè)務(wù)情況�����,制定人類遺傳資源活動(dòng)審批/ 備案�����、業(yè)務(wù)活動(dòng)風(fēng)險(xiǎn)等級(jí)劃分�����、不同風(fēng)險(xiǎn)等級(jí)業(yè)務(wù)審批流程�����、內(nèi)部稽查和自查�����、外部監(jiān)督檢查應(yīng)對(duì)等標(biāo)準(zhǔn)操作規(guī)程�����。還可以在標(biāo)準(zhǔn)操作規(guī)程基礎(chǔ)上制定體系化管理文件�����,以有效反映人類遺傳資源管理情況�����,如人類遺傳資源申請(qǐng)批準(zhǔn)進(jìn)度及問題跟蹤表�����、人類遺傳資源批件到期更新表�����、人類遺傳資源總結(jié)報(bào)告遞交進(jìn)度及問題跟蹤表、數(shù)據(jù)備份備案進(jìn)度及問題跟蹤表�����、自查及質(zhì)量事件報(bào)告進(jìn)展及問題跟蹤表�����、內(nèi)部稽查發(fā)現(xiàn)及整改計(jì)劃進(jìn)展跟蹤表�����、監(jiān)管機(jī)構(gòu)核查發(fā)現(xiàn)及整改計(jì)劃進(jìn)展跟蹤表等�����。
申辦者在人類遺傳資源合規(guī)管理制度建設(shè)過程中�����,應(yīng)及時(shí)關(guān)注人類遺傳資源相關(guān)法律法規(guī)和監(jiān)管部門官方問答的更新情況�����,并據(jù)此調(diào)整合規(guī)管理制度和體系化管理文件內(nèi)容�����。
四�����、結(jié)語(yǔ)
目前�����,我國(guó)臨床研究正處于高速發(fā)展階段�����,相關(guān)法律法規(guī)正根據(jù)實(shí)踐情況在不斷完善�����,監(jiān)管部門對(duì)于臨床研究也提出了越來越高的合規(guī)要求�����。由于臨床研究各參與方合規(guī)意識(shí)存在不足�����,并且臨床研究涉及的研究機(jī)構(gòu)、數(shù)據(jù)安全�����、人類遺傳資源等又分屬不同的監(jiān)管部門�����,所以不同監(jiān)管部門的聯(lián)合執(zhí)法�����、合規(guī)宣傳及培訓(xùn)顯得特別重要�����。申辦者作為臨床研究數(shù)據(jù)質(zhì)量和可靠性的最終責(zé)任人�����,應(yīng)結(jié)合實(shí)際情況和法律法規(guī)要求�����,選擇合適的臨床研究參與方并對(duì)其進(jìn)行監(jiān)督管理�����,遵守?cái)?shù)據(jù)合規(guī)和人類遺傳資源合規(guī)要求�����,建立體系化的臨床研究合規(guī)管理制度�����,使臨床研究合法合規(guī)開展�����,加快研究藥物上市進(jìn)度�����,造福廣大患者�����。