我們都知道�,F(xiàn)DA有權對生產FDA監(jiān)管產品的制藥公司進行突擊或預知檢查,并將其意見記錄在FDA 483表格上��,通?���?s寫為“483”。483 中引用的法規(guī)領域的觀察總結����,可以在 FDA 主頁的“檢查觀察Inspection Observations”小節(jié)中的電子表格獲得。除了觀察內容之外��,被引用的次數(shù)是這個表格透露的最直接的信息����。
藥物穩(wěn)定性研究是一門成熟的學科,當然也是一項重要的監(jiān)管要求����。然而在 2019 年至 2021 財年的許多 FDA 483 中都提到了穩(wěn)定性計劃的缺陷����。FDA 現(xiàn)已公布了 2022 財年(2021年10月至 2022年9月)的數(shù)據(jù)��。在“藥物”領域發(fā)布了 466 個 FDA 483�����。評估表明����,穩(wěn)定性計劃中的缺陷再次成為重災區(qū)���。
在“藥物”領域��,F(xiàn)DA過去幾年發(fā)布的報告數(shù)量如下:
2019 財年:779 份 FDA 483 份表格
2020 財年:349 份 FDA 483 份表格
2021 財年:215 份 FDA 483 份表格
2022 財年:466 份 FDA 483 份表格
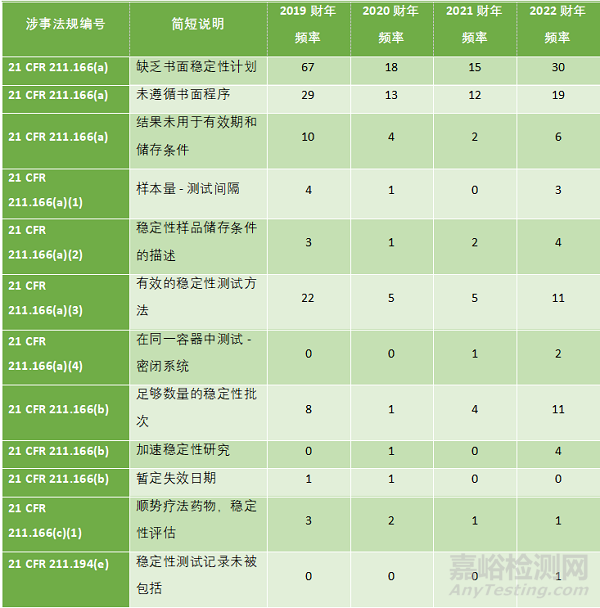
表1概述了與穩(wěn)定性測試相關的典型缺陷�。[1] 可以看出���,在上一財年的檢查中���,幾乎對 21 CFR Part 211.166 的所有子段落都有涉事項目�。
表1. FDA2022財年483表涉及穩(wěn)定性測試的觀察總結
二���、藥物穩(wěn)定性研究的監(jiān)管要求
穩(wěn)定性研究的要求在聯(lián)邦法規(guī)(Code of Federal Regulations)中有明確定義�����,主要體現(xiàn)在 21 CFR 211.166 條中:
1�����、211.166 穩(wěn)定性測試
(a) 應準備藥品穩(wěn)定性特征的書面測試程序��。這種穩(wěn)定性測試的結果應用于確定適當?shù)膬Υ鏃l件和有效期����。應遵循的書面計劃包括:
基于統(tǒng)計標準的樣本大小和測試間隔�����,用于檢查的每個屬性�,以確保對穩(wěn)定性的有效估計;
留樣保存條件��;
可靠、有意義��、具體的測試方法�;
在與上市藥品相同的容器密閉系統(tǒng)中對成品藥進行測試;
在規(guī)定配藥時間(按照標簽中的指示)檢測成品藥的重構(reconstitution)�����,以及在重構后進行檢測�。
(b) 應對每種藥品進行足夠數(shù)量的批次測試,以確定適當?shù)挠行?��,并保存此類?shù)據(jù)的記錄����。結合相關成分���、藥品和容器密閉系統(tǒng)所進行的加速研究,可用于支持暫定有效期�,但前提是完整的保質期研究結果暫不可完成,但正在進行中���。如果使用加速研究的數(shù)據(jù)來預測暫定有效期��,而預測的暫定有效期超出數(shù)據(jù)支持的實際保質期�����,則必須進行穩(wěn)定性研究���,包括在適當?shù)臅r間間隔進行藥品測試����,直到暫定有效期得到驗證或確定適合的有效期���。
(c) 對于順勢療法藥品的要求如下:
應至少基于對藥品成分相容性的測試或檢查���,并根據(jù)藥品的銷售經(jīng)驗,表明該藥品在正常情況下沒有降解��,對穩(wěn)定性進行書面評估或預估使用期限�����。
穩(wěn)定性評價應基于與上市藥品相同的容器密閉系統(tǒng)���。
(d) 標有“無美國效力標準No U.S. Standard of Potency”的過敏性提取物不受21 CFR 211.166要求的約束��。
另外����,21 CFR 211.194中也提到了穩(wěn)定性測試:
2、211.194 實驗室記錄
(e)應保存根據(jù) § 211.166 執(zhí)行的所有穩(wěn)定性測試的完整記錄�����。
三�����、藥物穩(wěn)定性測試要素
決定如何以及何時對新藥進行穩(wěn)定性測試����,是藥物研發(fā)和生命周期管理中的關鍵內容之一。穩(wěn)定性測試提供了原料藥或成品藥的質量與時間和保存環(huán)境之間的關聯(lián)�。它還規(guī)定了原料藥的復驗期、成品藥的保質期���,以及推薦的儲存條件。[3] FDA 的穩(wěn)定性測試要求與歐盟和日本協(xié)調并保持高度一致�����,因此在歐盟、日本或美國這三個地區(qū)中的任何一個地區(qū)生成的穩(wěn)定性信息將被其他兩個地區(qū)相互接受�����。
1��、原料藥穩(wěn)定性試驗的關鍵要素如下
(a) 壓力測試(stress testing):原料藥的壓力測試可以幫助識別可能的降解產物�,這反過來可以幫助建立降解途徑和分子的內在穩(wěn)定性,并驗證所用分析程序的穩(wěn)定性指示能力��。壓力測試的性質將取決于原料藥和涉及的成品藥類型�。
壓力測試可能對單批原料藥進行。它應包括溫度(以 10°C 的增量(例如����,50°C、60°C 等)高于加速測試的溫度)���、適當?shù)臐穸龋ɡ纾?5% RH 或更高)�����、氧化和對原料藥進行光解���。測試還應評估藥物物質在溶液或懸浮液中�����,在廣泛的 pH 值范圍內對水解的敏感性�����。光穩(wěn)定性測試應該是壓力測試的一個組成部分��。ICH Q1B 中描述了光穩(wěn)定性測試的標準條件�����。
在壓力條件下檢查降解產物���,有助于建立降解途徑,以及開發(fā)和驗證合適的分析程序���。但是�,如果已經(jīng)證明某些降解產物不是在加速或長期儲存條件下形成的�����,則可能沒有必要專門檢查它們��。
這些研究的結果將構成提供給監(jiān)管機構的信息的組成部分����。
(b) 批次的選擇:至少應使用三個primary批次(primary批次是指用于穩(wěn)定性研究的一批活性藥物成分或成品藥物,穩(wěn)定性數(shù)據(jù)在注冊申請中提交����,用于建立復驗期或保質期;primary批次的制造規(guī)模至少需要與中試(pilot scale)中的最小規(guī)模相當)的原料藥進行正式的穩(wěn)定性試驗�����。其生產過程必須與用于生產批次的最終過程相同�。進行正式穩(wěn)定性研究的原料藥批次的總體質量應能代表生產規(guī)模生產的原料藥的質量。
(c) 容器密閉系統(tǒng):必須對包裝在與最終包裝相同的容器密閉系統(tǒng)中的藥物進行測試�。
(d) 測試頻率:測試頻率應保證建立藥物穩(wěn)定性特征。對于建議復驗期至少為 12 個月的原料藥��,在長期貯存條件下的試驗頻率通常應為第一年每 3 個月一次���,第二年每 6 個月一次����,此后每年一次直至建議的復驗期����。
在加速儲存條件下��,在6 個月的研究框架下�����,建議至少三個時間點�����,包括初始和最終時間點(例如�����,0����、3 和 6 個月)�����。如果存在加速研究的結果可能接近顯著變化標準的預期(基于開發(fā)經(jīng)驗)��,則應通過在最后時間點添加樣本��,或通過在研究設計中包括第四個時間點來進行增加的測試。
(e) 當由于加速儲存條件發(fā)生顯著變化而要求在中期儲存條件下進行測試時�����,至少應該包括四個時間點���,包括初始和最終時間點(例如,0����、6、9�����、12 個月)���,從建議時間開始進行為期 12 個月的研究�。
(f) 存儲條件:測試必須評估存儲條件�����,以測試其熱穩(wěn)定性和對水分的敏感性��。這些條件還必須涵蓋儲存、運輸和后續(xù)使用�����。
(g) 規(guī)格Specification:ICH Q6A 和 Q6B 中闡述了規(guī)格�����,即測試內容����、分析方法和建議的驗收標準。此外�����,Q3A 中討論了原料藥中降解產物的規(guī)格�。穩(wěn)定性研究應包括對原料藥在儲存過程中容易發(fā)生變化,并對那些可能影響質量���、安全性和療效的那些屬性進行測試�����。測試應酌情涵蓋物理�、化學、生物學和微生物學屬性��。應采用經(jīng)過驗證的穩(wěn)定性分析程序�����。是否應該進行重復分析以及重復分析的程度取決于驗證研究的結果��。
2�、成品藥穩(wěn)定性試驗的關鍵要素如下
(a) 成品藥正式穩(wěn)定性研究應基于對原料藥行為和特性的了解�����,以及原料藥的穩(wěn)定性研究和臨床制劑研究中獲得的經(jīng)驗��。應說明在正式穩(wěn)定性研究中可能發(fā)生的儲存變化和選擇測試屬性的理由�����。
(b) 光穩(wěn)定性測試:如果合適���,應至少對一個初級批次的藥品進行光穩(wěn)定性測試����。ICH Q1B 中描述了光穩(wěn)定性測試的標準條件。
(c) 批次選擇:應提供至少三個primary批次藥品 (primary批次是指用于穩(wěn)定性研究的一批活性藥物成分或成品藥物�����,穩(wěn)定性數(shù)據(jù)在注冊申請中提交��,用于建立復驗期或保質期) 的穩(wěn)定性研究數(shù)據(jù)�����。primary批次應具有相同的配方�����,并包裝在與上市銷售相同的容器密閉系統(tǒng)中�。用于primary批次的制造過程應模擬生產批次的工藝,并應提供與銷售產品具有相同質量和符合相同規(guī)格的產品��。三批中的兩批至少應是中試規(guī)模批次�,如果有恰當理由,第三批可以小一些�。在可能的情況下,藥品的批次應使用不同批次的原料藥生產��。穩(wěn)定性研究應對藥品的每個單獨規(guī)格和容器尺寸進行,除非使用了涵括設計(bracketing)或矩陣設計(matrixing)��。
(d) 容器封閉系統(tǒng):穩(wěn)定性測試應在擬上市銷售的容器密閉系統(tǒng)中進行(包括任何二級包裝和容器標簽)��。
(e) 規(guī)格Specification:ICH Q6A 和 Q6B 中闡述了specification��,即測試列表����、分析程序參考和建議的驗收標準,包括放行和保質期規(guī)范的不同驗收標準的概念�����。此外�����,藥品中降解產物的規(guī)格在 Q3B 中進行了說明�����。
穩(wěn)定性研究應包括測試藥品在儲存過程中易發(fā)生變化并可能影響質量���、安全性和療效的那些屬性。測試應酌情包括物理、化學���、生物和微生物屬性���、防腐劑含量(例如抗氧化劑、抗菌防腐劑)和功能測試(例如劑量輸送系統(tǒng))�。分析程序應經(jīng)過充分驗證。是否應該進行重復分析以及重復分析的程度取決于驗證研究的結果�。
(f) 測試頻率:對于長期研究,測試頻率應足以確定藥品的穩(wěn)定性���。對于建議保質期至少為 12 個月的產品�,長期儲存條件下的測試頻率通常應為第一年每 3 個月一次����,第二年每 6 個月一次,此后在建議保質期內每年一次���。
在加速儲存條件下�,建議至少進行 6 個月的研究�,并至少涵蓋三個時間點,包括初始和最終時間點(例如�,0�、3 和 6 個月)���。如果預期(基于開發(fā)經(jīng)驗)加速測試的結果可能接近顯著變化標準�,則應通過在最后時間點添加樣本�,或通過在研究設計中包括第四個時間點來進行增加測試。
四����、藥物穩(wěn)定性研究條件
1、原料藥
(a) 常規(guī)條件存儲的原料藥
*由申請人決定是否在 25 ± 2°C/60% 相對濕度± 5% 相對濕度��,還是30°C ± 2°C/65% 相對濕度± 5%相對濕度下進行長期穩(wěn)定性研究�����。
*如果 30°C ± 2°C/65% 相對濕度± 5%相對濕度被選作長期條件�,則沒有中期研究�����。
如果選擇25°C ± 2°C/60% 相對濕度 ± 5% 相對濕度條件的長期研究���,并且在加速存儲條件下 6 個月測試期間的任何時間出現(xiàn)了“顯著變化”���,則應在中期存儲條件下進行額外測試���,并且應根據(jù)重大變更標準進行和評估。中期儲存條件下的測試應包括所有測試���,除非另有證明�����。初始申請應包括至少 6 個月的數(shù)據(jù)�����,這些數(shù)據(jù)來自在中間儲存條件下進行的為期 12 個月的研究����。原料藥的“顯著變化”被定義為不符合其質量標準(OOS)�����。
(b) 冰箱refrigerator內存儲的原料藥
如果在加速貯存條件下 3 到 6 個月的測試之間發(fā)生顯著變化�����,建議的復試期應基于長期貯存條件下可用的實時數(shù)據(jù)。如果在加速儲存條件下的前 3 個月測試中發(fā)生顯著變化���,則應提供討論,以解決標簽儲存條件之外的短期偏移的影響����,例如在運輸或處理過程中。如果合適�,可以通過對單個批次的原料藥進行短于 3 個月、但比平時更頻繁的測試�,進一步測試來支持這種討論。如果在前 3 個月內發(fā)生了顯著變化�����,則沒有必要繼續(xù)測試原料藥 6 個月�����。
(c) 擬冷藏在freezer中的原料藥
對于擬在freezer中貯存的原料藥����,復驗期應基于在長期貯存條件下獲得的實時數(shù)據(jù)����。對于擬儲存freezer中的原料藥���,在沒有加速儲存條件的情況下,應在升高的溫度(例如 5°C ± 3°C 或 25°C ± 2°C)環(huán)境下��,對單個批次進行適當?shù)臏y試�,以確定短期偏離建議標簽存儲條件的時間段,例如在運輸或處理過程中�。
(d) 擬在 -20°C 以下儲存的原料藥
擬在 -20°C 以下儲存的原料應根據(jù)具體情況進行處理。
2���、成品藥
(a) 常規(guī)條件存儲的成品藥
*由申請人決定是否在 25 ± 2°C/60% 相對濕度± 5% 相對濕度���,還是30°C ± 2°C/65% 相對濕度± 5%相對濕度下進行長期穩(wěn)定性研究。
*如果 30°C ± 2°C/65% 相對濕度± 5%相對濕度被選作長期條件�,則沒有中間條件。
如果在 25°C ± 2°C/60% RH ± 5% RH 下進行長期研究��,并且在加速存儲條件下 6 個月測試期間的任何時間出現(xiàn)“顯著變化”�,中期存儲條件下進行的額外測試應根據(jù)重大變更標準進行和評估。初始申請應包括至少 6 個月的數(shù)據(jù)��,這些數(shù)據(jù)來自在中期儲存條件下進行的為期 12 個月的研究�����。
一般來說,藥品的“顯著變化”定義為:
測定值較初始值有 5% 的變化�;或在使用生物或免疫程序時未能達到效力的驗收標準;
降解產物超標���;
未能滿足外觀�����、物理屬性和功能測試的驗收標準(例如��,顏色�����、相分離�、再懸浮性���、結塊�����、硬度����、每次啟動的劑量輸送)�����;然而��,在加速條件下可能會發(fā)生一些物理屬性的變化(例如�����,栓劑軟化���,乳膏融化)����;
pH值不符合驗收標準�����;
12個劑量單位溶出度不合格�。
(b) 擬儲存在冰箱refrigerator中的成品藥
如果藥品包裝在半透性容器中,應提供適當?shù)男畔⒁栽u估失水程度。
如果在加速儲存條件下 3 至 6 個月的測試中發(fā)生顯著變化�,則建議的保質期應基于長期儲存條件下可用的實時數(shù)據(jù)。如果在加速儲存條件下的前 3 個月測試中發(fā)生顯著變化���,則應提供討論����,以解決標簽儲存條件之外的短期偏移的影響�����,例如在運輸和處理過程中�����。如果合適���,可以通過在短于 3 個月��、但比平時更頻繁的測試期間��,對單批藥品進行進一步測試來支持這種討論�。如果在前 3 個月內發(fā)生了顯著變化����,則沒有必要繼續(xù)測試產品 至6 個月����。
(c) 擬冷藏于freezer的成品藥
對于擬在freezer中儲存的藥品�����,保質期應基于在長期儲存條件下獲得的實時數(shù)據(jù)�����。對于擬儲存在freezer中的藥品�����,在沒有加速儲存條件的情況下�����,應在高溫(例如 5°C ± 3°C 或 25°C ± 2°C)下對單個批次進行適當?shù)臏y試��,并確定一個時間段�,以解決在建議的標簽存儲條件之外的短期偏移的影響�。
(d) 擬在 -20°C 以下儲存的成品藥
擬在 -20°C 以下儲存的藥品應根據(jù)具體情況進行處理���。
參考文獻:
[1] ECA Academy. In 2022 again Numerous FDA 483s due to Deficiencies in the Stability Program. 07. 12. 2022
[2] ICH. ICH Harmonised Tripartite Guideline Stability Testing of New Drug Substances and Products Q1A(R2). 06. 02. 2003.
[3] FDA (2003) Guidance for Industry Q1A(R2) Stability Testing of New Drug Substances and Products retrieved on 05/06/2021 from https://www.fda.gov/media/71707/download