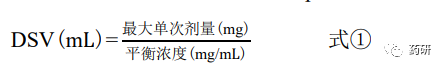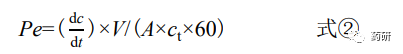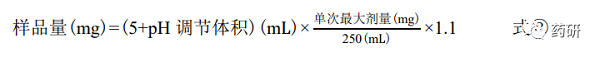摘要:測定了磷酸奧司他韋(2)和索非布韋(3)的平衡溶解度、油水分配系數(shù)和滲透性����。采用 搖瓶法,分別測定了 2 和 3 在 pH 1.2�、pH 4.5 及 pH 6.8 緩沖液中的平衡溶解度����,判斷藥物的 溶解性�;測定了 2 和 3 在不同 pH 條件下的正辛醇緩沖液體系中的油水分配系數(shù);并通過平 行人工膜體外滲透技術(shù)��,測定了 2 和 3 在 pH 5.0��、pH 6.5 及 pH 7.4 緩沖液中的滲透性�。結(jié)果 顯示,2 為高溶解性�、高滲透性藥物,油水分配系數(shù)受油水體積比影響較大��;3 為低溶解性��、 高滲透性藥物��,具有較高的脂溶性�。然而,用相同方法測定甲基多巴(1)時(shí)��,由于樣品穩(wěn)定性 的問題����,該藥物的溶解性未得出結(jié)論����。此外��,本研究闡述并分析了研究過程中可能出現(xiàn)的問 題和影響因素��,為 WHO 基本藥物目錄的建立提供了數(shù)據(jù)支持��。
人體生物等效性試驗(yàn)豁免適用于仿制藥質(zhì)量和療效一致性評價(jià)中口服固體常釋制劑申請 生物等效性(bioequivalence����,BE)豁免[1—3]�,以國際公認(rèn)的生物藥劑學(xué)分類系統(tǒng)(BCS)為依據(jù) [4]。而藥物溶解性�、腸道滲透性以及制劑溶出度是 BCS 系統(tǒng)的 3 個(gè)關(guān)鍵因素,其中高溶解性 藥物可作為固體口服制劑 BE 豁免的重要指標(biāo)[5]����。
世界衛(wèi)生組織(WHO)于 2009 年公布了基本藥物目錄中的 BCS 分類,2011 年美國 FDA 也總結(jié)歸納了一套 BCS 分類[6]�。但是,由于 WHO 和美國 FDA 公布的 BCS 分類數(shù)據(jù)主要來 源于文獻(xiàn)收集����,未經(jīng)過試驗(yàn)驗(yàn)證��。因此��,WHO 于 2016 年啟動(dòng)了 BCS 分類實(shí)驗(yàn)室驗(yàn)證項(xiàng)目 [7]��,由于化合物品種繁多����,共分配給全球 9 個(gè)實(shí)驗(yàn)室(包括本研究單位)進(jìn)行�,本研究于 2019~ 2021 年完成了其中具有代表性的 3 個(gè)品種,即分別對甲基多巴(methyldopa��,1)����、磷酸奧司他 韋(oseltamivir phosphate,2)以及索非布韋(sofosbuvir�,3)3 種藥物的平衡溶解度、油水分配系 數(shù)以及滲透性進(jìn)行了研究[8]�。1~3 均被納入 WHO 基本藥物目錄中,化合物結(jié)構(gòu)式見圖 1�。
1 于 1960 年首次被發(fā)現(xiàn),是一種?2 受體機(jī)動(dòng)型心血管藥物�,臨床主要用于治療高血壓����、 妊娠型高血壓和先兆子癇��。1 片最早由日本米諾源發(fā)制藥株式會(huì)社研發(fā)并獲批上市����,單次最 大劑量為 250 mg��。2 是神經(jīng)氨酸酶抑制劑�,即抗病毒類藥物,2 膠囊(商品名為達(dá)菲)于 1999 年被美國 FDA 批準(zhǔn)上市��,并于 2004 年 7 月在中國上市����,單次最大劑量為 75 mg。3 是美國吉 利德科學(xué)公司研發(fā)的治療慢性丙肝的新藥�,該藥物于 2013 年經(jīng)美國 FDA 批準(zhǔn)上市,于 2014 年經(jīng)歐洲藥品管理局(EMA)批準(zhǔn)在歐盟各國上市�,還未在中國上市,單次最大劑量為 400 mg����。
目前平衡溶解度的測定方法主要有雙指示劑滴定法、電位滴定法和酸堿滴定法等[9—10]。本研究采用搖瓶法和高效液相色譜法(HPLC)法測定藥物的平衡溶解度��,并通過藥物溶解性與 滲透性評價(jià)系統(tǒng)(平行人工膜滲透技術(shù)����,PAMPA[11] )對 3 種藥物的油水分配系數(shù)及滲透性進(jìn) 行了測定,同時(shí)對研究過程中發(fā)現(xiàn)的問題進(jìn)行了分析與討論����,確定了藥物的 BCS 分類,以期為基本藥物目錄的制定提供了數(shù)據(jù)支持����。
一、儀器與試藥
e2695 型高效液相色譜系統(tǒng)和 2489 型 UV/Vis 檢測器(美國 Waters 公司)��;ME155DU 型電子天平和 S470-K 型 pH 計(jì)(瑞士 Mettler Toledo 公司)�;TURBISCAN TOWER 型多重光散射儀-穩(wěn)定性分析儀(法國 Formulaction 公司);µDISS Profiler 藥物溶解性與滲透性評價(jià)系統(tǒng)(美國 Pion 公司)�;聚四氟乙烯濾膜(美國 安捷倫公司,0.45 μm�,13 mm);離心管(德國 Eppendorf 公司��,聚丙烯�,25 mL)�。
1(匈牙利 Egis Pharmaceuticals PLC 公司�,含量 99.8%,批號 600301217)�、2(印度 Laurus Labs Limited 公司,含量 99.7%��,批號 AOTV-2/VSP1/004/18)��、3(印度 Solara Active Pharma Sciences Limited 公司�,含量 99.5%,批號 PL40047113)�,以上樣品均由 WHO 委托國外企業(yè)生產(chǎn)����;2 對照品(含量 99.8%,批號 101096- 200901)和維生素 C(含量 100%��,批號 100425-201103)(中國食品藥品檢定研究院)����;胃腸道模擬脂質(zhì)體(批 號 520807)和 ASB 緩沖液(acceptor sink conditioned buffer,批號 520825)(美國 Pion 公司)�;無水乙酸鈉(美 國 Sigma 公司,含量 99.0%��,批號 BCCB2261);正辛醇�、氯化鈉、二水合磷酸二氫鈉��、磷酸鈉��、磷酸二氫 鉀��、氫氧化鉀��、氫氧化鈉����、鹽酸、冰乙酸和磷酸均為分析純����,甲醇和乙腈為色譜純,試驗(yàn)所用溶液均用電 阻率為 18.2 MΩ·cm 的去離子水配制����。
二、方法與結(jié)果
2.1 HPLC 法測定藥物的溶解性及方法學(xué)驗(yàn)證
2.1.1 色譜條件
1:色譜柱 Waters Symmetry® C18柱(4.6 mm×250 mm��,5 μm)����;流動(dòng)相 甲醇∶乙腈∶pH 6.0磷酸鹽緩沖液(稱取磷酸二氫鉀6.8 g����,加水980 mL使其溶解����,用1 mol/L氫氧化鉀溶液調(diào)至 pH 6.0)(245∶135∶620);流速1.2mL/min��;柱溫50 ℃�;檢測波長207 nm;進(jìn)樣量15μL����。
2:色譜柱 Agilent XBridge C8柱(4.6 mm×250 mm����,5 μm);流動(dòng)相 甲醇∶0.1 mol/L pH 3.0磷酸鈉緩沖液(稱取磷酸鈉16.4 g�,加水1 L使其溶解,用磷酸溶液調(diào)至pH 3.0)(15∶85)����;流速1.0 mL/min��;柱溫35 ℃�;檢測波長280 nm�;進(jìn)樣量20 μL。
3:色譜柱 Agilent XDB C18柱(4.6 mm×250 mm��,5 μm)��;流動(dòng)相 乙腈∶水(30∶70)����;流 速 1.0mL/min;柱溫35 ℃��;檢測波長260 nm��;進(jìn)樣量20 μL����。
2.1.2 溶液配制
pH 5.0、pH 6.5及pH 7.4緩沖液:均參照《中華人民共和國藥典》2020年版(ChP 2020)四 部通則8004緩沖液項(xiàng)下配制����。
標(biāo)準(zhǔn)曲線溶液:精密稱取1(由于WHO提供的樣品量較大,且純度較高�,因此用樣品配制 標(biāo)準(zhǔn)曲線溶液)100 mg�,置50 mL量瓶中����,加入0.1 mol/L鹽酸溶液使其溶解并定容,作為1標(biāo)準(zhǔn) 曲線貯備液����,精密量取1標(biāo)準(zhǔn)曲線貯備液適量,用0.1 mol/L鹽酸溶液稀釋成每1 mL中含1 0.2��、 0.4�、0.8、1.0����、2.0 mg的1標(biāo)準(zhǔn)曲線溶液。精密稱取2對照品1 g��,置100 mL量瓶中����,加入稀釋劑 [水∶甲醇∶乙腈(620∶245∶135)]使其溶解并定容��,作為2標(biāo)準(zhǔn)曲線貯備液��,精密量取2標(biāo)準(zhǔn)曲線貯備液適量,用上述稀釋劑制成每1 mL中含2 1��、2�、3、4�、5 mg的2標(biāo)準(zhǔn)曲線溶液。精密 稱取3 10 mg�,置20 mL量瓶中,加入乙腈∶水(30∶70)使其溶解并定容����,作為3標(biāo)準(zhǔn)曲線貯備 液,精密量取3標(biāo)準(zhǔn)曲線貯備液適量����,用乙腈∶水(30∶70)制成每1 mL中含3 5.09、10.18��、20.36����、 40.72、50.90 μg的3標(biāo)準(zhǔn)曲線溶液��。
質(zhì)控樣品溶液:取1,精密稱定����,加入0.1 mol/L鹽酸溶液使其溶解并稀釋成質(zhì)量濃度約為 0.8 mg/mL的溶液,平行配制2份�,其中1份經(jīng)0.45 μm濾膜過濾,取續(xù)濾液及未過濾的溶液作為 1質(zhì)控樣品溶液����。取2對照品,精密稱定����,加入稀釋劑[水∶甲醇∶乙腈(620∶245∶135)]使其 溶解并稀釋成質(zhì)量濃度約為3.0 mg/mL的溶液,平行配制2份��,其中1份經(jīng)0.45 μm濾膜過濾�,取 續(xù)濾液及未過濾的溶液作為2質(zhì)控樣品溶液。取3�,精密稱定,加入乙腈∶水(30∶70)使其溶 解并稀釋成質(zhì)量濃度約為0.2 mg/mL的溶液�,平行配制2份,其中1份經(jīng)0.45 μm濾膜過濾�,取續(xù) 濾液及未過濾的溶液作為3質(zhì)控樣品溶液。
2.1.3 系統(tǒng)適用性試驗(yàn)
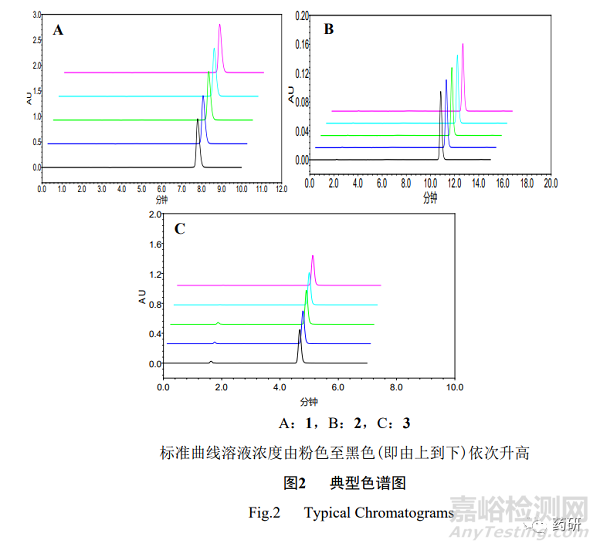
分別取“2.1.2”項(xiàng)下 1~3 標(biāo)準(zhǔn)曲線溶液�,分別按“2.1.1”項(xiàng)下方法進(jìn)樣測定,結(jié)果見圖 2�。1~3 峰理論塔板數(shù)分別為 13 026、10 733��、10 312����,拖尾因子分別為 1.15、1.32����、1.12;標(biāo) 準(zhǔn)曲線溶液(中間濃度點(diǎn))連續(xù)進(jìn)樣 5 針的峰面積 RSD(n=5)分別為 0.05%�、0.11%、0.17%����。
2.1.4 線性試驗(yàn)
分別取“2.1.2”項(xiàng)下的 1~3 標(biāo)準(zhǔn)曲線溶液,分別按“2.1.1”項(xiàng)下色譜條件進(jìn)樣測定����,以 質(zhì)量濃度 c 為橫坐標(biāo),峰面積 A 為縱坐標(biāo)進(jìn)行線性回歸��。結(jié)果顯示����,1~3 分別在 0.20~2.00 mg/mL�、1.001~5.005 mg/mL��、5.09~50.9 μg/mL 內(nèi)線性關(guān)系良好��,回歸方程分別為 A=13 842 307c+231 497��,r=0.999 9�;A=13 609 650c+4 951 215,r=0.998 7��;A=10 703c+3 554��,r=0.999 7�。
2.1.5 精密度試驗(yàn)
分別取“2.1.2”項(xiàng)下的 1~3 標(biāo)準(zhǔn)曲線溶液(中間濃度點(diǎn)),分別按“2.1.1”項(xiàng)下色譜條件進(jìn)樣 測定����,連續(xù)進(jìn)樣 5 針。結(jié)果顯示 1~3 峰面積的 RSD(n=5)均小于 0.2%��,說明方法精密度較高��。
2.1.6 準(zhǔn)確度試驗(yàn)
分別取“2.1.2”項(xiàng)下的 1~3 質(zhì)控樣品溶液����,分別按“2.1.1”項(xiàng)下色譜條件進(jìn)樣測定��,以 標(biāo)準(zhǔn)曲線法計(jì)算過膜后的溶液溶度。結(jié)果顯示��,1~3 質(zhì)控樣品溶液過膜前后質(zhì)量濃度的誤差 均小于 1.0%����,回收率在 99.1%~100.0%。
2.1.7 回收率試驗(yàn)
分別取“2.1.2”項(xiàng)下的 1~3 質(zhì)控樣品溶液�,分別加入等體積“2.1.2”項(xiàng)下的 1~3 標(biāo)準(zhǔn) 曲線溶液,分別配制成低����、中、高 3 個(gè)濃度水平的加標(biāo)供試品溶液��,按“2.1.1”項(xiàng)下色譜條 件進(jìn)樣測定并計(jì)算回收率�。結(jié)果顯示,1~3 的平均回收率(n=3)分別為 99.69%����、99.90%、 99.46%�,RSD 分別為 0.04%�、0.04%�、0.05%。
2.2 平衡溶解度的測定
2.2.1 緩沖液的配制
pH 1.2 緩沖液:稱取氯化鈉 2.52 g�,加入水 900 mL 使其溶解,用 70 g/L 的鹽酸溶液調(diào)至 pH 1.2��,再用水定容至 1 000 mL��。
pH 4.5 緩沖液:稱取乙酸鈉 2.99 g�,加入水 900 mL 使其溶解,用 120 g/L 乙酸溶液調(diào)至 pH 4.5��,再用水定容至 1 000 mL����。
pH 4.5 緩沖液:稱取乙酸鈉 2.99 g,加入水 900 mL 使其溶解�,用 120 g/L 乙酸溶液調(diào)至 pH 4.5,再用水定容至 1 000 mL�。
2.2.2 測定方法及結(jié)果
平衡溶解度的測定采用搖瓶法,稱取 1~3 樣品適量�,分別加入“2.2.1”項(xiàng)下的 3 種 pH 值的緩沖液適量,形成過飽和溶液(固體樣品過量 10%~30%)��,并用 0.1 mol/L 鹽酸溶液或 0.1 mol/L 氫氧化鈉溶液調(diào)節(jié) pH 值至初始值附近��,每個(gè) pH 值平行配制 3 份。置恒溫?fù)u床中��, 轉(zhuǎn)速 120 r/min��,溫度(37±0.5)℃����,分別在 2����、4、6��、8�、12、24����、48、72 h 取樣 1 mL����,用 0.45 μm 濾膜過濾,棄去初濾液��,取續(xù)濾液適量,用對應(yīng) pH 值緩沖液進(jìn)行定量稀釋�,使質(zhì)量濃度 在標(biāo)準(zhǔn)曲線范圍內(nèi),若超出范圍可進(jìn)行再次稀釋��,并按“2.1.1”項(xiàng)下色譜條件進(jìn)樣測定��。
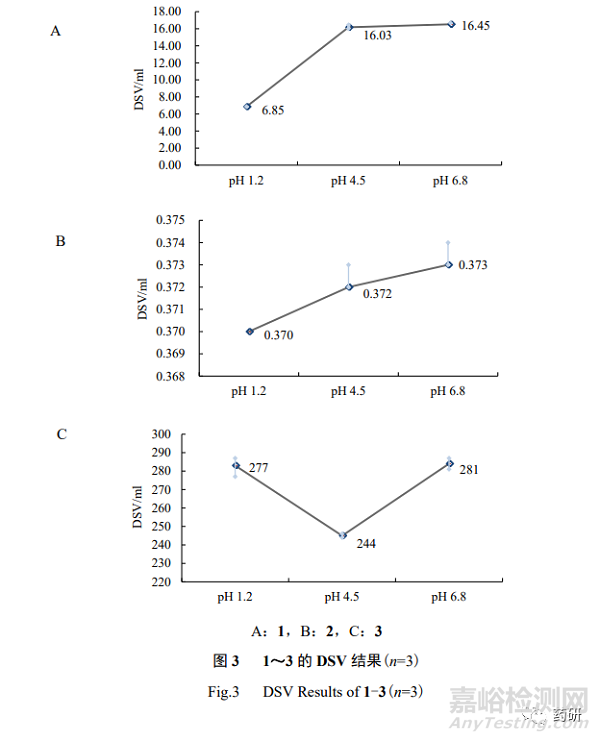
根據(jù)不同時(shí)間點(diǎn)取樣的 HPLC 測定濃度值����,繪制時(shí)間濃度曲線,平臺(tái)濃度點(diǎn)(相鄰 2 個(gè)取 樣點(diǎn)濃度相近)即為“平衡時(shí)間”����,該時(shí)間點(diǎn)取樣的濃度即為“平衡溶解度”。此外�,按式① 分別計(jì)算不同 pH 值緩沖液的溶解度體積(DSV,mL)����,當(dāng)所有 pH 值條件下的 DSV 均低于 250 mL,則判斷為高溶解性����;若某 pH 值條件下的 DSV 高于 250 mL,則判斷為低溶解性。
1~3 的 DSV 結(jié)果見圖 3����,綜合 3 個(gè) pH 值條件下的 DSV 結(jié)果,2 為高溶解性��;3 為低 溶解性��,而 1 的溶解性為無法判斷(無法判斷的原因詳見本研究討論部分“3.1”項(xiàng)下)����。
2.3 油水分配系數(shù)的測定
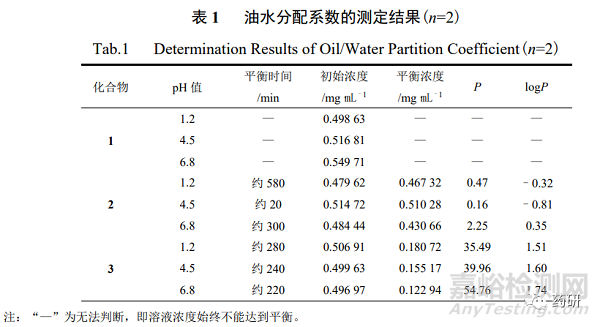
采用藥物溶解性與滲透性評價(jià)系統(tǒng)測定油水分配系數(shù),分別稱取 1~3 樣品適量�,分別用 “2.2.1”項(xiàng)下不同 pH 緩沖液配制成 1~3 質(zhì)量濃度各約為 0.5 mg/mL 的溶液�,分別量取上述 溶液 18 mL,置 25 mL 水浴池中����,分別加入由相應(yīng) pH 緩沖液飽和的正辛醇溶液 1 mL,內(nèi)置 轉(zhuǎn)子(150 r/min)�,于 37 ℃水浴鍋中 24 h,光纖探頭置于緩沖液層�。1~3 的檢測波長分別為 205~210 nm、275~285 nm�、255~265 nm,每分鐘采集 1 次����,待溶質(zhì)分配平衡時(shí)分別計(jì)算油 相(o)和水相(w)的質(zhì)量濃度(co����、cw)����,并計(jì)算油水分配系數(shù)(P,co/cw)及 logP�,結(jié)果見表 1。
綜合平衡溶解度與油水分配系數(shù)的測定結(jié)果可知����,2 的水溶性較強(qiáng),其表觀分配系數(shù)受 水相組成�、緩沖液 pH 值和油水體積比的影響不大;3 則具有較強(qiáng)的脂溶性����,在 3 種不同的 pH 緩沖液中均得到較高的油水分配系數(shù);在 3 種 pH 緩沖液中均無法測定 1 的油水分配系數(shù)����, 24 h 內(nèi)水相(w)中 1 濃度無明顯變化。有研究表明,油水分配系數(shù)過低�,藥物不易透過脂質(zhì)分 子膜,油水分配系數(shù)過高��,藥物因強(qiáng)脂溶性難以進(jìn)入淋巴�、血液系統(tǒng)中,無法發(fā)揮藥效�,為 了讓藥物得到充分吸收,使其發(fā)揮最佳藥效�,藥物的油水分配系數(shù)最好在–2~3 內(nèi)[12—14]。
2.4 滲透性的測定
采用PAMPA技術(shù)測定1~3樣品的滲透性��,稱取1~3樣品約4 mg�,置藥物溶解性與滲透性 評價(jià)系統(tǒng)的供體室,精密量取“2.2.1”項(xiàng)下不同pH緩沖液和ASB緩沖液各20 mL��,分別置供體 室和受體室中��,兩室間以面積為1.65 cm2的仿生膜(經(jīng)脂質(zhì)體22 μL浸潤)隔離����,溫度為37 ℃��, 攪拌速度為150 r/min��,采集時(shí)間為8 h,采樣間隔60 s��,檢測波長為220~320 nm����,采用光纖探 頭分別實(shí)時(shí)監(jiān)測藥物濃度,按照式②計(jì)算藥物的有效滲透性(Pe����,cm/s)。根據(jù)美國FDA發(fā)布 的《固體口服制劑的體內(nèi)生物利用度和生物等效性研究》指導(dǎo)原則以及其給出的不同滲透性 藥物[15]����,本研究分別選取了高滲透性藥物,即美托洛爾和普洛萘爾�,在同等條件下測定其Pe值以建立模型,通過對比結(jié)果來判斷2和3的滲透性��。
式中 Pe 為有效滲透性(cm/s)�;ct 為供體室中藥物的初始質(zhì)量濃度(μg/mL);dc/dt 為單位 時(shí)間內(nèi)受體室中藥物濃度的變化速率(μg·mL–1·min–1)�;V 為受體室緩沖液的體積(mL);A 為 膜面積(cm2)��。
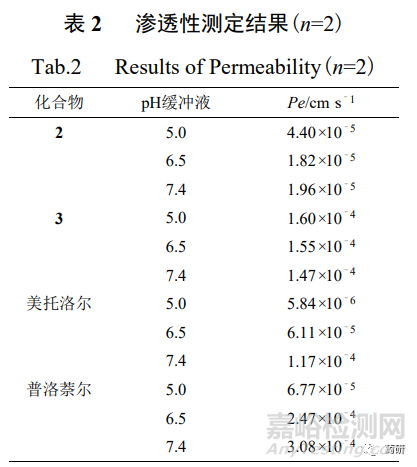
2 和 3 的滲透性測定結(jié)果見表 2(1 是主動(dòng)轉(zhuǎn)運(yùn)機(jī)制[16]�,通過該系統(tǒng)測定其滲透性無實(shí)際 意義)����,2 和 3 在不同 pH 緩沖液體系中的 Pe 值基本均大于模型藥物(2 在 pH 7.4 緩沖液體系 除外)�,可判斷 2 種藥物均為高滲透性藥物。
三����、 討論
3.1 藥物穩(wěn)定性對溶解度判斷結(jié)果的影響
在整個(gè)研究過程中發(fā)現(xiàn),2 和 3 在 3 個(gè)不同 pH 值緩沖液中較穩(wěn)定��,1 在 pH 6.8 緩沖液中 會(huì)發(fā)生降解�,在試驗(yàn)過程中溶液會(huì)由無色逐漸變?yōu)榉凵罱K變?yōu)楹谏?�,并伴有黑色沉淀產(chǎn) 生�。由于整個(gè)試驗(yàn)過程中溶液始終保持過飽和狀態(tài),因此�,通過 HPLC 測定仍然能夠找到溶 解平衡點(diǎn)。通過多重光散射儀對溶液的透光率進(jìn)行分析��,發(fā)現(xiàn) 1 在 pH 6.8 緩沖液中溶液的透 光率在 16 h 內(nèi)從 0 降至約 25%����;在同樣的條件下�,適當(dāng)加入維生素 C��,發(fā)現(xiàn)溶液的透光率未 發(fā)生變化����。通過穩(wěn)定性研究以及查閱相關(guān)文獻(xiàn)得知[17]�,1 在中性或堿性條件下會(huì)發(fā)生氧化反 應(yīng),產(chǎn)生黑色沉淀�。從色譜分析數(shù)據(jù)以及計(jì)算結(jié)果來看,1 在 3 個(gè) pH 緩沖液條件下的平均 DSV 值均小于 250 mL��,應(yīng)當(dāng)判斷為高溶解性����。然而,此次研究是由國際多個(gè)實(shí)驗(yàn)室同時(shí)進(jìn)行 比對分析��,對于 1 藥物��,國內(nèi)��、國際 3 個(gè)實(shí)驗(yàn)室得出了不同的結(jié)論��,因此對于該藥物的溶解 性目前仍未給出結(jié)論����。
3.2 調(diào)節(jié) pH 對溶解度試驗(yàn)的影響
測定平衡溶解度時(shí)�,在藥物穩(wěn)定的基礎(chǔ)上�,pH 值的調(diào)節(jié)也尤為重要。2 和 3 在 3 個(gè)緩沖 液中����,pH 值均能調(diào)至初始 pH 值附近。1 在 pH 1.2 緩沖液條件下配制溶液時(shí)�,依據(jù) 1 單次最 大劑量為 250 mg,試驗(yàn)時(shí)首先稱取 1 約 300 mg��,加入 pH 1.2 緩沖液 10 mL�,振搖至全部溶 解,再次加入 1 100 mg����,發(fā)現(xiàn)少量樣品未溶解,測定 pH 值約為 1.8�,采用 0.1 mol/L 鹽酸溶液 調(diào)至 pH 1.2,發(fā)現(xiàn)所有樣品全部溶解��,再次加入樣品 100 mg��,再次發(fā)生上述現(xiàn)象����。因此�,本 次研究不再繼續(xù)調(diào)節(jié) pH 值�,使供試品溶液始終保持過飽和狀態(tài)�。
3.3 樣品量及緩沖液體積的確定
在整個(gè)研究過程中,供試品溶液始終維持在過飽和狀態(tài)�,對于高溶解性藥物來說,對樣 品量的要求較高�。例如在本次研究中,WHO 提供的 3 僅為 0.640 g��,這對試驗(yàn)設(shè)計(jì)提出了較 高的要求�。本次研究采用不同 pH 值的緩沖液 5 mL,依據(jù)單次最高劑量分別計(jì)算每個(gè) pH 值 條件下的所需樣品量(見式③)��,從而保證試驗(yàn)結(jié)果的準(zhǔn)確性�。
3.4 滲透性測定中波長段的選擇
以 3 在 pH 6.5 緩沖液體系中的滲透性測定為例,供體室中樣品濃度從 4 h 起趨于平衡����, 故本試驗(yàn)節(jié)選 4~8 h 的滲透數(shù)據(jù)進(jìn)行統(tǒng)計(jì)計(jì)算,選取紫外吸收圖譜中下坡段曲線�,即 290~ 300 nm 波長段進(jìn)行實(shí)時(shí)監(jiān)測。
3.5 BCS 分類
基于上述研究結(jié)果可知�,2 為高溶解性-高滲透性藥物,BCS 分類應(yīng)為 1 類��;3 為低溶解 性-高滲透性藥物,BCS 分類應(yīng)為 2 類��。
WHO 基本藥物目錄在不斷地補(bǔ)充與更新�,目前沒有公認(rèn)的方法對藥物平衡溶解度進(jìn)行 測定。此外����,藥物的 BCS 分類也急需試驗(yàn)數(shù)據(jù)作為支撐。本研究通過測定 3 種不同藥物的平 衡溶解度����、油水分配系數(shù)以及滲透性,闡述并分析了研究過程中可能出現(xiàn)的問題以及試驗(yàn)結(jié) 果的影響因素����,為 WHO 基本藥物目錄的建立提供了數(shù)據(jù)支持。