強(qiáng)降解試驗的目的并非是為了實現(xiàn)分析結(jié)果的質(zhì)量平衡��,而是對降解化學(xué)有一個比較全面的了解:1)了解藥物的降解路徑及分子內(nèi)穩(wěn)定性���;2)建立穩(wěn)定性指示分析方法,使其適用于樣品檢測���;3)為藥品的處方�����、工藝��、包裝���、貯藏條件的確定提供有益支持,以便于穩(wěn)定性試驗的順利進(jìn)行�����。在一些情況下���,降解產(chǎn)物(雜質(zhì))質(zhì)量(或摩爾數(shù))的增加小于母體化合物(主成分)質(zhì)量(或摩爾數(shù))相應(yīng)的減少�����。問題的潛在來源和解決方法如下所述:
(一)降解產(chǎn)物在色譜柱上未被洗脫
假定母體化合物和所有降解產(chǎn)物都是完全可溶的��,并且可以通過HPLC-UV檢測���。有以下方法可以診斷此問題:
(a)可以修改HPLC方法以洗脫其他雜質(zhì)
可以通過增加流動相的強(qiáng)度(即增大有機(jī)相比例)或增加分析時間來修改HPLC方法,以洗脫保留較強(qiáng)的非極性化合物���。
(b)可以使用紫外分光光度法將部分降解的樣品與未降解進(jìn)行分析�����,并將結(jié)果與通過HPLC分析獲得的結(jié)果進(jìn)行比較���。
此方法對于使用UV檢測器的HPLC方法很有用。由于紫外分光光度法不涉及分離�����,因此不會由于化合物保留在色譜柱上而被遺漏。使用這種方法��,可以將部分降解的樣品在流動相中溶解或稀釋���。獲得部分降解樣品完整的UV(或VIS)光譜���,并將其與未降解樣品的光譜進(jìn)行比較。在HPLC方法中使用的波長下獲得部分降解樣品與未降解樣品的吸光度之比��。然后將該吸光度比與通過HPLC方法獲得的部分降解樣品與未降解樣品的總峰面積之比進(jìn)行比較���。如果使用HPLC方法檢測到所有雜質(zhì)���,則部分降解樣品的總HPLC峰面積除以未降解樣品的HPLC峰面積應(yīng)等于吸光度比。如果HPLC方法利用光電二極管陣列(PDA)檢測器���,則可以根據(jù)需要在多個波長下確定比較�����。如果HPLC面積比明顯小于分光光度法吸光度比��,則存在一種或多種降解產(chǎn)物沒有從色譜柱上洗脫��。
(c)可以使用不接色譜柱的HPLC系統(tǒng)分析樣品(即流動注射分析)�����。
在該實驗中���,用雙通代替HPLC系統(tǒng)中的色譜柱,將獲得的總峰面積與使用色譜柱時獲得的總峰面積進(jìn)行比較��。如果在接色譜柱時獲得的總峰面積明顯小于不接色譜柱時的總峰面積���,則HPLC方法中存在一些雜質(zhì)未被洗脫���。在不接色譜柱的情況下,所有雜質(zhì)和主成分共同洗脫��,得到一個未保留的峰�����,其面積與可檢測物質(zhì)的總量成正比例�����。假設(shè)所有物質(zhì)都被洗脫,則色譜柱存在時所有峰的總峰面積應(yīng)相同��。如果色譜柱存在時與不接色譜柱時總峰面積差異較大��,則在給定的色譜條件下很可能一種或多種化合物沒有被洗脫��。這種診斷方式的潛在困難是易受溶解樣品溶劑的影響�����,若溶劑與流動相有很大不同�����,則溶劑對樣品響應(yīng)的影響可能會使在沒有色譜柱的情況下難以進(jìn)行準(zhǔn)確的積分���。此外���,進(jìn)樣量產(chǎn)生的峰面積不能超出線性范圍。最后�����,如果在使用色譜柱時使用了流動相梯度�����,則不斷變化的溶劑組成會影響分析物的響應(yīng),從而影響洗脫物質(zhì)的峰面積���。當(dāng)梯度流動相組成的每個極端值均等地使用時(等度),可以通過比較不使用色譜柱而獲得的總面積來確定梯度是否具有這種影響�����。
(二)檢測器未檢測到降解產(chǎn)物
HPLC-UV是最常用的檢測技術(shù)�����。盡管廣泛適用���,但紫外檢測器不能檢測所有化合物��。降解可能會產(chǎn)生沒有生色團(tuán)的化合物�����,在這種情況下���,觀察到的降解產(chǎn)物增加量將小于母體化合物的損失量��。該問題的診斷可使用更低的波長或使用其他檢測器(例如���,蒸發(fā)光散射檢測(ELSD),帶電氣溶膠檢測(CAD)�����,質(zhì)譜法(MS)或火焰電離檢測(FID)��。必須牢記的是�����,雖然此類檢測器適用范圍廣���,但與UV一樣��,其響應(yīng)也不均勻���。例如,蒸氣壓明顯的化合物通過ELSD和CAD給出的響應(yīng)較差�����,而MS響應(yīng)隨可電離性變化很大。但是這種檢測器對于確認(rèn)原始方法未檢測到的降解產(chǎn)物的存在可能非常有用���。
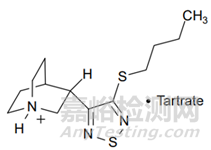
檢測器未檢測到降解產(chǎn)物的示例如候選藥物L(fēng)Y297802�����。LY297802水溶液在冷白色熒光燈(約17000 lux)下破壞3天�����,質(zhì)量損失了5.4%;破壞7天���,質(zhì)量損失了42%�����。但是�����,通過HPLC-UV有關(guān)物質(zhì)方法(梯度方法)��,降解產(chǎn)物的增加量分別為0.4%和1.3%���,明顯存在分析質(zhì)量不平衡��。仔細(xì)檢查盛有樣品的容器���,發(fā)現(xiàn)表面沉積了不溶性霧狀薄膜。收集該不溶性薄膜并通過EI-MS分析��,表明該薄膜含有元素硫�����。由于硫很可能源自噻二唑環(huán)的降解���,考慮生成沒有生色團(tuán)化合物的可能性�����,及形成揮發(fā)性產(chǎn)物的可能性�����。用己烷萃?����。◤妮p度降解的水溶液中堿化��,得到LY297802的游離堿)�����,然后進(jìn)行GC-FID分析��,發(fā)現(xiàn)降解樣品中有兩種主要的降解產(chǎn)物���。使用GC-MS進(jìn)行分析���,提供了分子式信息���,闡明了結(jié)構(gòu)���,提供了對降解化學(xué)的了解以及HPLC-UV缺乏可檢測性的原因(如,降解產(chǎn)物易揮發(fā)且無紫外吸收)�����。
圖1 LY297802結(jié)構(gòu)
在某些情況下��,由于不溶性,揮發(fā)性或吸附損失���,降解產(chǎn)物會無意中從檢測樣品中排除�����。通常���,最難解決的問題是最明顯且最直接的解決方法。在這種情況下�����,目視觀察或濁度檢測可能會發(fā)現(xiàn)問題所在���。例如�����,在較早描述的化合物L(fēng)Y297802的降解溶液中��,觀察到一些不溶性物質(zhì)���,并確定其為元素硫��,為質(zhì)量平衡問題提供了有價值的線索���。當(dāng)然,當(dāng)存在于含有不溶性輔料的藥品中時��,不溶性降解產(chǎn)物不太明顯��。在這種情況下��,與安慰劑或未降解的樣品相比��,不溶性化合物的分離和檢查是合適的���。
如果降解產(chǎn)物由于其揮發(fā)性而損失��,則可以使用其他分析技術(shù)�����。在這種情況下,使用溶劑-溶劑提取樣品可能是合適的���。萃?�。ㄈ缜懊嫣岬降腖Y297802示例中所述)或以頂空方式捕獲降解樣品���。然后可以使用氣相色譜法(GC-FID或GC-MS)比較降解和未降解樣品的結(jié)果�����。在有限的情況下��,如果降解是由熱引起且迅速發(fā)生��,則TGA和相關(guān)的氣相色譜分析可能是一種有用的方法���。最后,如果降解可以在低溫下產(chǎn)生��,則可以使樣品保持低溫以最小化揮發(fā)性���。
降解產(chǎn)物也可能吸附在樣品容器或不溶性輔料中��,在后一種情況下��,問題的診斷和解決方法與降解產(chǎn)物不溶性相同(參見上文)���。如果吸附到容器上是一個潛在的問題��,那么最直接的方法是使用不同的容器材料(例如玻璃和聚丙烯)比較結(jié)果���。在某些情況下,可能需要對容器表面進(jìn)行去活或更改樣品溶劑(例如pH和溶劑強(qiáng)度)以最大程度地減少吸附���。
(三)母體化合物在樣品基質(zhì)中丟失
在極少數(shù)情況下��,由于揮發(fā)性或吸附性��,母體化合物本身可能會從樣品基質(zhì)中流失�����。通常���,即使發(fā)生此類損失,與母體化合物的質(zhì)量相比也微不足道���。但是���,如果有意義的話,測定結(jié)果的下降將不會是由于降解引起的��,因此不會對應(yīng)于降解產(chǎn)物的任何增加�����。通常���,在強(qiáng)降解試驗之前獲得的信息(例如���,熔點、沸點�����,蒸氣壓和吸附到各種材料上的趨勢)應(yīng)提供此類潛在問題的跡象���。如上所述進(jìn)行診斷和解決�����。
(四)在有關(guān)物質(zhì)方法中與母體化合物共洗脫的降解產(chǎn)物
如果在有關(guān)物質(zhì)方法中降解產(chǎn)物與母體化合物共洗脫�����,那么在給定條件下�����,所得質(zhì)量平衡取決于雜質(zhì)相對于母體化合物的響應(yīng)因子�����。如果所形成的雜質(zhì)具有較小的響應(yīng)因子�����,則存在質(zhì)量平衡為正�����。如果雜質(zhì)的響應(yīng)因子大于母體的響應(yīng)因子�����,那么質(zhì)量平衡將為負(fù)���。如果響應(yīng)因子相同�����,則不會影響質(zhì)量平衡�����。通過光電二極管陣列(PDA)-紫外檢測或通過MS檢測可以揭示共洗脫雜質(zhì)為峰異質(zhì)性���。當(dāng)然,PDA檢測僅對具有不同UV光譜且不能與母體完美共洗脫的雜質(zhì)有效���。另外��,PDA對檢測峰異質(zhì)性的敏感性取決于雜質(zhì)的光譜特性與母體有多大差異���。對于某些雜質(zhì),PDA檢測器可能無法檢測到含量低于百分之幾的雜質(zhì)��。LC-MS可以對廣泛的潛在共洗脫雜質(zhì)提供更高的靈敏度�����。然而�����,LC-MS更昂貴,可以使用的流動相類型有限�����。也可使用正交分離技術(shù)(例如�����,CE)來檢查共洗脫雜質(zhì)�����。如果發(fā)現(xiàn)共洗脫問題���,應(yīng)修改有關(guān)物質(zhì)方法以分離出共同洗脫的雜質(zhì)���。
(五)色譜性能不良導(dǎo)致降解產(chǎn)物未積分
一些降解產(chǎn)物的色譜性能可能不佳例如由于與痕量金屬雜質(zhì)或殘留硅醇基的不良相互作用,在柱上從一種產(chǎn)物轉(zhuǎn)化為另一種產(chǎn)物等���,盡管它們可能從HPLC色譜柱上洗脫下來��,但所得色譜峰變寬了(通常嚴(yán)重拖尾)��,很容易被“遺漏”并保持未積分狀態(tài)�����,尤其是當(dāng)寬峰且峰面積較低時���。如果母體化合物降解為溶解性差的雜質(zhì)���,則分析后觀察到的色譜圖可能不會顯示出離散的峰�����,而是基線升高�����。在低水平下�����,這樣高的基線很容易被遺漏�����。諸如此類的情況并不少見,特別是在使用等度HPLC方法的情況下��。運行空白對于確定基線升高是來自樣品還是屬于色譜假象��,非常有幫助���。確定質(zhì)量平衡結(jié)果是否來自不良的色譜性能的實驗與確定降解產(chǎn)物是否未從色譜柱中洗脫的實驗相同(請參見“未從HPLC色譜柱中洗脫的降解產(chǎn)物”一節(jié))��。
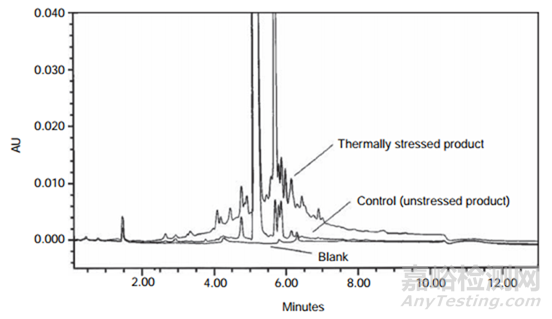
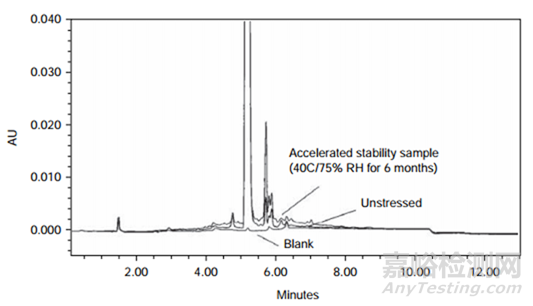
母體化合物降解產(chǎn)物之間分離度較差的示例如圖1所示�����。在本例中�����,通過RP-HPLC分析了部分降解的樣品(在25分鐘內(nèi)�����,乙腈的梯度為5-70%)���,并且檢測到許多具有已知響應(yīng)因子的已知降解產(chǎn)物,但是質(zhì)量平衡為88.6%��。考慮了降解產(chǎn)物未從色譜柱上洗脫下來的可能性���,并設(shè)計了一個實驗來檢驗該假設(shè)�����。在該實驗中��,在沒有接色譜柱的情況下分析了降解樣品(流動注射)���。如果使用RP-HPLC方法檢測到所有降解產(chǎn)物���,則HPLC含量和有關(guān)物質(zhì)結(jié)果的總和(即83.5 + 5.1 = 88.6%)應(yīng)等于流動注射結(jié)果��。流動注射結(jié)果為97.3%���,表明使用RP-HPLC方法未檢測到大量的降解產(chǎn)物。該差異與降解產(chǎn)物未從色譜柱上洗脫出來或未進(jìn)行積分相一致��。為了研究產(chǎn)物是否未從色譜柱上洗脫���,對梯度RP-HPLC方法進(jìn)行了改進(jìn)��,將乙腈的比例提高至90%��,并在此比例下保持1小時��,但并未洗脫出任何其他降解產(chǎn)物��,這表明沒有任何強(qiáng)保留的非極性降解產(chǎn)物�����。為了測試降解產(chǎn)物色譜性能不佳的可能性���,進(jìn)行了另一項實驗�����。在該實驗中��,使用等度HPLC方法對樣品進(jìn)行了測定���,該方法有機(jī)相比例較高,以單峰洗脫母體化合物和降解產(chǎn)物�����。獲得的結(jié)果與流動注射分析的結(jié)果相當(dāng),這表明所有降解產(chǎn)物都從色譜柱上洗脫下來���。該發(fā)現(xiàn)表明色譜分離不良的降解產(chǎn)物未與原始RP-HPLC方法積分在一起���。通過將原始RP-HPLC梯度修改為陡峭梯度(在25分鐘內(nèi)使用0-90%乙腈),證實了這一假設(shè)��。如圖3所示��,比較了未破壞樣品和降解樣品(質(zhì)量平衡較差)�����。注意基線大約在3-8分鐘升高�����,僅在破壞樣品上可見�����,而在對照樣品上看不到�����。將該基線“峰”的積分并包括在總峰面積(母體和總雜)中���,得到的結(jié)果與使用流動注射分析獲得的結(jié)果相似�����。當(dāng)色譜圖和空白的色譜圖重疊時���,在降解不那么嚴(yán)重的樣品中也可以識別出該基線峰(如圖4所示)。因此���,低的質(zhì)量平衡歸因于色譜性能差的物種���,這些雜質(zhì)以前沒有被積分,因為它們被忽略為僅僅是基線波動�����。
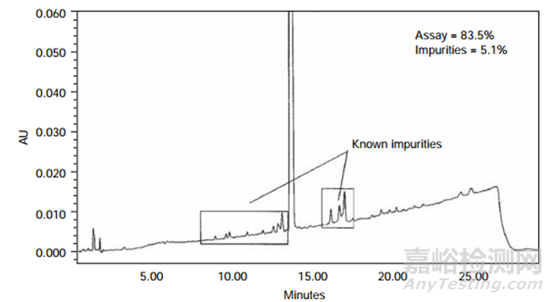
圖2 使用梯度HPLC在高溫破壞樣品上獲得的HPLC色譜圖
圖3使用陡梯度HPLC方法在高溫破壞樣品�����,未破壞樣品和空白樣品上獲得的HPLC色譜圖疊加圖
圖4 使用陡梯度HPLC方法分析6個月加速穩(wěn)定性條件下的樣品��,未破壞的樣品和空白所獲得的疊加HPLC色譜圖
(六)響應(yīng)因子差異導(dǎo)致定量不準(zhǔn)確
如果未校正的峰面積代表實際相對量���,雜質(zhì)和母體化合物之間的響應(yīng)因子(例如��,給定波長下的吸收率)顯著不同���,則質(zhì)量不守恒是不可避免的。如果降解產(chǎn)物的響應(yīng)因子小于母體化合物的響應(yīng)因子���,則會導(dǎo)致質(zhì)量平衡為正��。當(dāng)降解產(chǎn)物具有較大的響應(yīng)因子時��,將導(dǎo)致質(zhì)量平衡為負(fù)��。因此��,雜質(zhì)的相對響應(yīng)因子(RRF)是評估質(zhì)量平衡時的重要考慮因素�����。
有時,可以合理地假設(shè)UV響應(yīng)因子非常相似�����。例如,如果母體和降解產(chǎn)物共享相同的生色主鏈�����,則僅在與生色團(tuán)無關(guān)的區(qū)域具有結(jié)構(gòu)差異�����。UV光譜的有利比較(例如���,來自PDA檢測器的光譜)可以幫助確認(rèn)這種情況�����。
通常�����,建立響應(yīng)因子的過程需要使用單個雜質(zhì)的對照品���。為了準(zhǔn)確測定,必須知道雜質(zhì)的純度��,然后根據(jù)峰面積及濃度比計算響應(yīng)因子。但是要綜合考慮雜質(zhì)與母體化合物之間的紫外吸收光譜圖��,選擇合適的紫外吸收波長�����,否則���,也會導(dǎo)致質(zhì)量不守恒�����。
因此��,在強(qiáng)制降解試驗前需考慮樣品的理化質(zhì)�����,了解藥物在各個條件下可能的降解機(jī)制以及降解反應(yīng)的難易程度�����,可能形成的降解產(chǎn)物�����,合理的設(shè)計試驗��,以確定合適的降解條件��。
參考文獻(xiàn):
[1] Pharmaceutical Stress Testing Predicting Drug DegradationSecond Edition.Steven W.Baertschi,Karen M. Alsante,Robert A. Reed�����。