2022年已經(jīng)過去,在這一年中美國CDER一共批準(zhǔn)37款創(chuàng)新藥��,其中包含22個新分子實(shí)體和15個新生物制品申請��,這也是2017年以來批準(zhǔn)新藥數(shù)量最少的一年��。批準(zhǔn)的藥物中��,抗癌新藥依舊占主體��,多個重磅抗癌療法陸續(xù)上市,給患者帶來了更多選擇��。
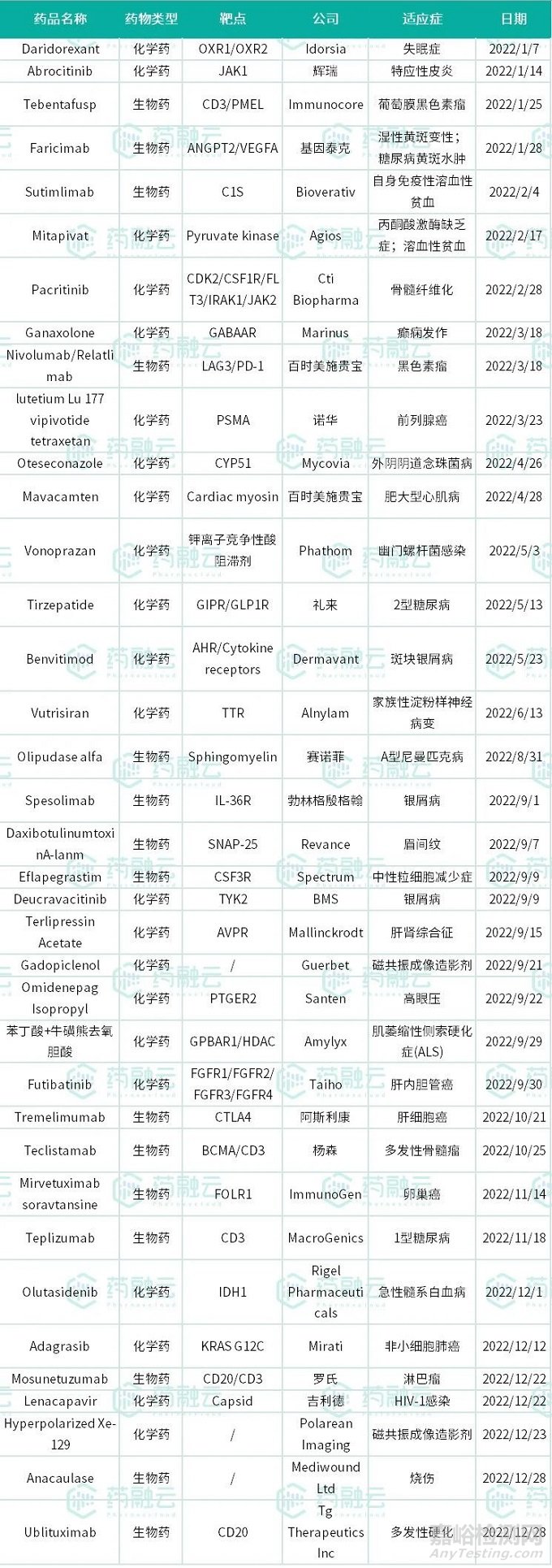
數(shù)據(jù)來源:藥融云企業(yè)版數(shù)據(jù)庫綜合查詢
1��、Quviviq(Daridorexant)
“Quviviq”(通用名:Daridorexant)為一款治療失眠癥的藥物��,它是一種雙重食欲素受體拮抗劑(OXR)��。治療機(jī)制是通過競爭性結(jié)合OXR1和OXR2��,抑制食欲素信號功能從而阻斷覺醒通路以抑制過度活躍的失眠狀態(tài)。該項批準(zhǔn)是基于3期臨床試驗��,共入組1854名成年人,結(jié)果顯示與安慰劑組相比��,藥物組在入睡時間��、睡眠持續(xù)時間以及總睡眠時間都有顯著改善。
2��、Cibinqo(Abrocitinib)
“Cibinqo”(Abrocitinib)��,是一種口服JAK抑制劑��,用于治療現(xiàn)有的全身療法控制不充分的患有復(fù)發(fā)性中度至重度特應(yīng)性皮炎的成人患者。臨床3期結(jié)果顯示:與安慰劑相比��,藥物組在疾病程度��、疾病嚴(yán)重性及皮膚清潔程度上均有明顯改善��,且患者能在給藥治療兩周后瘙癢癥狀得到快速緩解。該藥在2021年已經(jīng)在日本和歐洲獲得了批準(zhǔn)��。
3��、Kimmtrak(Tebentafusp)
“Kimmtrak”(Tebentafusp)是融合T細(xì)胞受體和抗CD3免疫效應(yīng)結(jié)構(gòu)域的雙特異性融合蛋白療法��。以TCR技術(shù)為基礎(chǔ)��,通過靶向黑色素細(xì)胞和惡性黑色素瘤表達(dá)的gp100為目標(biāo),實(shí)現(xiàn)對癌細(xì)胞的靶向性殺傷��。該藥是美國首個被批準(zhǔn)用于治療“HLA-A*02:01陽性不可切除或轉(zhuǎn)移性葡萄膜黑色素瘤”的藥物也是首款獲得FDA監(jiān)管批準(zhǔn)的T細(xì)胞受體(TCR)療法��。
4��、Vabysmo(Faricimab)
“Vabysmo”(Faricimab)是一款治療新生血管(濕性)年齡相關(guān)黃斑退化(nAMD)和糖尿病性黃斑水腫(DME)患者的可注射眼用藥物��。該藥為首款獲FDA批準(zhǔn)的眼科領(lǐng)域雙特異性抗體療法��,通過選擇性的阻斷VEGF-A和血管生成素-2(Ang-2)兩條關(guān)鍵致病通路達(dá)到治療疾病的作用。在臨床試驗中��,約半數(shù)受試者的注射間隔延長至4個月,與當(dāng)前標(biāo)準(zhǔn)治療方法相比(間隔2個月注射的aflibercept)��,視力獲益達(dá)到非劣效性標(biāo)準(zhǔn)��。
5、Enjaymo(Sutimlimab)
“Enjaymo”(sutimlimab)是FDA批準(zhǔn)的首款治療冷凝集素病的單克隆抗體��,劑型為注射劑��,通過抑制紅細(xì)胞的破壞(溶血),降低病人因溶血而致的紅細(xì)胞(RBC)輸血需求��。該項批準(zhǔn)是基于臨床III期試驗結(jié)果:Enjaymo達(dá)到了主要療效終點(diǎn)��,54%的患者達(dá)到主要復(fù)合終點(diǎn)��。該品曾獲得孤兒藥��、突破療法和優(yōu)先審評資格��,目前在日本已經(jīng)獲批上市,歐盟新藥上市申請正在進(jìn)行中��。
6��、Pyrukynd(Mitapivat)
2022年2月17日,F(xiàn)DA批準(zhǔn)了Agios的mitapivat用于治療丙酮酸激酶(PK)缺乏的成人溶血性貧血(一種紅細(xì)胞破壞速度大于生成速度的疾?�。T撍帿@得孤兒藥資格��、快速通道和優(yōu)先審查資格��。
Mitapivat的有效性在兩項研究中進(jìn)行了評估��。一項是隨機(jī)、雙盲��、安慰劑對照的臨床研究��,研究對象為80例未接受常規(guī)輸血的成年P(guān)KD患者��。另一項是單臂研究,研究對象為27例接受定期輸血的成年P(guān)KD患者��。
在隨機(jī)研究中��,mitapivat的有效性是基于血紅蛋白反應(yīng)��,定義為研究開始時血紅蛋白濃度增加≥1.5 g/dL,并在≥2次評估中保持穩(wěn)定��。血紅蛋白濃度是用于測量紅細(xì)胞未被破壞的數(shù)量��。在研究結(jié)束時��,40%接受mitapivat治療的患者出現(xiàn)血紅蛋白反應(yīng)��,而接受安慰劑的患者中無人出現(xiàn)血紅蛋白反應(yīng)。
在單臂研究中��,mitapivat有效性基于輸血負(fù)擔(dān)的減少��,定義為在過去24周的治療中��,與患者的既往輸血負(fù)擔(dān)相比至少減少33%的紅細(xì)胞單位。33%接受mitapivat治療的患者實(shí)現(xiàn)了輸血負(fù)擔(dān)的減少��,其中22%的參與者在過去24周的治療中不需要任何輸血��。
7��、Vonjo(Pacritinib)
2022年02月28日CTI BioPharma Corp.宣布美國食品藥品監(jiān)督管理局(FDA)批準(zhǔn)VONJO(pacritinib)用于治療患有中度或高危原發(fā)性的成人或繼發(fā)性(真性紅細(xì)胞增多癥或原發(fā)性血小板增多癥后)骨髓纖維化。
加速批準(zhǔn)上市是基于VONJO在骨髓纖維化患者中的關(guān)鍵3期PERSIST-2研究的療效結(jié)果?�;颊甙?:1:1隨機(jī)分配接受VONJO 200 mg每日兩次(BID)��、VONJO 400 mg每日一次(QD)或最佳可用療法(BAT)。允許先前的JAK2抑制劑治療��。
在這項研究中��,在基線血小板計數(shù)低于50×10 9的患者隊列中/L接受pacritinib 200 mg BD治療的患者中��,29%的患者脾臟體積減少至少35%,而接受包括蘆可替尼(ruxolitinib)在內(nèi)的最佳可用療法的患者中有3%的患者脾臟體積減少了至少35%��。作為加速批準(zhǔn)的一部分��,CTI需要在驗證性試驗中描述臨床益處��。為了滿足這一批準(zhǔn)后的要求,CTI計劃完成PACIFICA試驗��,預(yù)計在2025年年中取得結(jié)果��。
8��、Ztalmy(Ganaxolone)
2022年03月18日,Marinus Pharmaceuticals公司宣布��,美國FDA已批準(zhǔn)Ztalmy(ganaxolone)加奈索酮口服混懸劑用于治療在2歲以上的患者中與細(xì)胞周期蛋白依賴性激酶樣5(CDKL5)缺乏癥(CDD)相關(guān)的癲癇發(fā)作��。Ztalmy是首款獲得FDA批準(zhǔn)針對CDD患者群體的療法��。這一批準(zhǔn)是基于一項隨機(jī)雙盲,含安慰劑對照的3期臨床試驗數(shù)據(jù)��,共入組101例患者��。在治療第28天時��,試驗達(dá)到了主要終點(diǎn),Ztalmy組患者主要運(yùn)動癲癇發(fā)作頻率的中位降低幅度為30.7%(p=0.0036)��,安慰劑組為降低6.9%��。在開放標(biāo)簽擴(kuò)展研究中��,接受Ztalmy治療至少12個月的患者(n=48)��,主要運(yùn)動癲癇發(fā)作頻率中位降低幅度為49.6%。安全性上��,該3期試驗中��,Ztalmy通常耐受良好��,并顯示與既往臨床試驗一致的安全性特征��,最常見的不良事件為嗜睡。
9��、Opdualag(Nivolumab/Relatlimab)
2022年03月18日��,百時美施貴寶宣布美國FDA批準(zhǔn)其First-in-class雙免疫療法Relatlimab+Nivolumab固定劑量組合Opdualag上市,治療罹患不可切除或轉(zhuǎn)移性黑色素瘤的成人和兒童(12歲及以上)患者��。Relatlimab是美國FDA批準(zhǔn)的首款LAG-3抗體��,也是近10年來針對全新免疫檢查點(diǎn)獲批的首款創(chuàng)新癌癥免疫療法��。FDA本次對relatlimab的批準(zhǔn)主要基于隨機(jī)��、雙盲的Ⅱ/Ⅲ期臨床試驗RELATIVITY-047��。RELATIVITY-047(CA224-047)是一項隨機(jī)、雙盲��、II/III期研究��,用于評估relatlimab聯(lián)用Opdivo相較于Opdivo單藥治療既往未經(jīng)治療的轉(zhuǎn)移性或不可切除的黑色素瘤患者。
試驗主要終點(diǎn)是無進(jìn)展生存期(PFS)��,次要終點(diǎn)是總生存期(OS)和客觀緩解率(ORR)��。共有714例患者按1:1的比例隨機(jī)接受relatlimab(160 mg)+Opdivo(480 mg)vs Opdivo(480 mg)進(jìn)行靜脈輸注,每4周一次��,直至疾病復(fù)發(fā)��、出現(xiàn)不可接受的毒性或撤回知情同意��。目前正在對次要終點(diǎn)OS和ORR進(jìn)行隨訪��。Opdualag™(nivolumab and relatlimab-rmbw)每次注射價格為$27,389在療效方面��,Relatlimab–Opdivo組的中位無進(jìn)展生存期為10.1個月,而Opdivo組為4.6個月��。Relatlimab–Opdivo組的12個月PFS率為47.7%��,而Opdivo組為36.0%��。在安全性方面,Relatlimab–Opdivo組和Opdivo組分別有18.9%和9.7%的患者發(fā)生3級或4級治療相關(guān)不良事件��。
10��、Pluvicto(Lutetium Lu 177 vipivotide tetraxetan)
2022年03月23日��,諾華宣布,美國FDA已經(jīng)批準(zhǔn)該公司的靶向放射性配體療法Pluvicto(lutetium Lu 177 vipivotide tetraxetan)上市用于治療前列腺特異性膜抗原(PSMA)陽性轉(zhuǎn)移性去勢抵抗性前列腺癌(mCRPC)患者��,這些患者先前已經(jīng)接受過基于紫杉烷的化療和雄激素受體信號通路抑制劑治療��。
這一批準(zhǔn)是基于一項關(guān)鍵性3期臨床試驗的積極結(jié)果��。試驗結(jié)果顯示��,與標(biāo)準(zhǔn)治療相比��,添加Pluvicto將患者的死亡風(fēng)險降低38%��,Pluvicto同時顯著降低患者出現(xiàn)放射學(xué)疾病進(jìn)展或死亡的風(fēng)險。而且��,在基線攜帶可評估疾病的患者中��,Pluvicto組的總緩解率為30%,標(biāo)準(zhǔn)治療對照組這一數(shù)值為2%��。
11��、Vivjoa(Oteseconazole)
2022年04月26日,F(xiàn)DA官網(wǎng)顯示FDA已批準(zhǔn)Mycovia Pharmaceuticals公司開發(fā)的口服抗真菌藥物Vivjoa(oteseconazole)用于在不具有生殖潛力的女性中��,降低復(fù)發(fā)性外陰陰道念珠菌?�。≧VVC)的復(fù)發(fā)幾率��。
這一批準(zhǔn)是基于3項3期臨床試驗的積極結(jié)果的支持��,包括2項全球性的VIOLET試驗和1項在美國進(jìn)行的ultraVIOLET試驗��,一共入組870多例患者。兩項VIOLET試驗均達(dá)到其主要終點(diǎn)和關(guān)鍵次要終點(diǎn)��。在為期48周的試驗中��,超過90%的患者接受oteseconazole治療后能夠有效預(yù)防感染復(fù)發(fā)(p<0.001)��。
此外,ultraVIOLET試驗也達(dá)到其所有主要終點(diǎn)與關(guān)鍵次要終點(diǎn)��。試驗結(jié)果表明��,oteseconazole治療VVC初始發(fā)作有效,并與目前標(biāo)準(zhǔn)治療相比��,進(jìn)一步證明了其治療RVVC的療效和安全性��。到50周時��,治療組復(fù)發(fā)率為5.1%��,對照組為42.2%(p<0.001)��。并且��,oteseconazole可保護(hù)95%的受試者近一年不復(fù)發(fā)。期待這款新藥能夠為女性患者帶來新的治療選擇��。
12��、Camzyos(Mavacamten)
2022年04月28日(新澤西州普林斯頓),百時美施貴寶(BMS)公司宣布��,其“first-in-class”心肌肌球蛋白別構(gòu)抑制劑Camzyo(mavacamten)獲得美國FDA批準(zhǔn)��,用于治療梗阻性肥厚型心肌病(oHCM)成人患者��。
這一批準(zhǔn)是基于關(guān)鍵性3期臨床試驗EXPLORER-HCM的結(jié)果��,EXPLORER-HCM 3期試驗是一項雙盲��、隨機(jī)��、安慰劑對照、平行組試驗��,共招募了251名有癥狀(NYHA II或III級)阻塞性肥厚性心肌病的成年患者��。
EXPLORER-HCM的主要終點(diǎn)是復(fù)合功能終點(diǎn)��,在30周評估��,定義為混合靜脈氧分壓(pVO 2)改善≥1.5 mL/kg/min的患者比例NYHA分級至少1級或pVO 2提高≥3.0 mL/kg/min且NYHA分級無惡化。與安慰劑組相比��,Camzyos組在第30周達(dá)到主要終點(diǎn)的患者比例更高(分別為37%和17%��,差異為19%(95%CI:9��、30;p=0.0005)��。在第30周��,與安慰劑組相比��,接受Camzyos的患者在所有次要終點(diǎn)方面都有更大的改善��。
13��、Voquezna(Vonoprazan/Amoxicillin/Clarithromycin;伏諾拉生+阿莫西林+克拉霉素)
2022年05月03日Phathom宣布美國食品和藥物管理局(FDA))批準(zhǔn)了VOQUEZNA™TRIPLE PAK™(vonoprazan,amoxicillin,clarithromycin)伏諾拉生片劑��、阿莫西林膠囊��、克拉霉素片劑和VOQUEZNA™DUAL PAK™(vonoprazan,amoxicillin)伏諾拉生片劑、阿莫西林膠囊用于治療成人幽門螺桿菌感染��。
這些批準(zhǔn)基于PHALCON-HP 3期試驗的安全性和有效性數(shù)據(jù)��,這是美國有史以來在幽門螺桿菌中進(jìn)行的最大的注冊試驗,隨機(jī)分配了1,046名患者��。在改良的意向治療人群中��,兩種VOQUEZNA治療方案在基線時無克拉霉素或阿莫西林耐藥性幽門螺桿菌菌株的患者中均表現(xiàn)出不劣于蘭索拉唑三聯(lián)療法��。
14��、Mounjaro(Tirzepatide)
2022年05月13日,禮來宣布��,美國FDA批準(zhǔn)該公司葡萄糖依賴性促胰島素多肽(GIP)和胰高血糖素樣肽-1(GLP-1)受體雙重激動劑Mounjaro(tirzepatide)替西帕肽注射劑上市��,每周注射1次��,輔助飲食和運(yùn)動,以改善成人2型糖尿病患者的血糖控制��。
此次批準(zhǔn)是基于III期SURPASS的項目積極結(jié)果��,其大型III期項目由10項臨床試驗組成��,計劃招募超過13000例2型糖尿病患者��,其中5項試驗是全球性的注冊研究。其中包括與司美格魯肽1 mg��、甘精胰島素和德谷胰島素的活性對照研究��。這些研究評估了Mounjaro(5mg10mg和15mg)單獨(dú)使用或與常用的糖尿病處方藥物(包括二甲雙胍��、SGLT2抑制劑��、磺脲類藥物和甘精胰島素)聯(lián)合使用的療效。SURPASS項目中��,Mounjaro 5mg劑量平均使受試者A1C減少1.8%-2.1%��,10mg和15mg劑量平均使受試者A1C減少了1.7%-2.4%��。
此外��,Tirzepatide在減重方面也取得了成功,在III期SURMOUNT-1試驗中��,tirzepatide(5mg,10mg,15mg)治療組患者第72周時的減重效果均顯著優(yōu)于安慰劑對照組��,平均減重最高達(dá)到22.5%(24kg)��,而且15mg高劑量組有63%的患者減重達(dá)到20%以上。禮來表示將與FDA討論并尋求這一適應(yīng)癥的監(jiān)管批準(zhǔn)��。
15��、Vtama(Benvitimod)
2022年05月24日,Dermavant Sciences公司宣布��,美國FDA已批準(zhǔn)Vtama(1%tapinarof)本維莫德每日1次乳膏用于外用治療斑塊狀銀屑病成人患者��。這一批準(zhǔn)包括所有銀屑病患者��,不論嚴(yán)重程度如何��,并且標(biāo)簽上沒有使用持續(xù)時間和使用身體部位的限制。
在兩項關(guān)鍵性3期臨床試驗中��,tapinarof達(dá)到所有的主要和次要終點(diǎn)��,名為PSOARING 1的臨床試驗中,36%接受tapinarof治療的患者達(dá)到皮膚癥狀清除或接近清除的標(biāo)準(zhǔn)��,對照組這一數(shù)值為6%��。在另一項3期臨床試驗中��,這兩個數(shù)值分別為40%和6%(p<0.0001)��。在長期擴(kuò)展研究中��,獲得皮膚癥狀完全清除的患者在停藥之后可平均維持皮膚光潔或接近光潔4個月��。為期52周的長期療效數(shù)據(jù)顯示,接受tapinarof乳膏治療的成人斑塊狀銀屑病患者��,在療效結(jié)果��、生活質(zhì)量方面均獲得持久改善��,未出現(xiàn)療法失效的情況。長期擴(kuò)展試驗的患者滿意度數(shù)據(jù)顯示��,81.7%的患者認(rèn)為這款療法比以前用過的外用療法更為有效��。
16��、Amvuttra(Vutrisiran)
2022年06月13日��,Alnylam公司宣布��,美國FDA批準(zhǔn)首款皮下注射RNAi療法Amvuttra(vutrisiran)上市��,用于治療遺傳性轉(zhuǎn)甲狀腺素蛋白介導(dǎo)(hATTR)的淀粉樣變性成人患者的多發(fā)性神經(jīng)?�。╬olyneuropathy)。FDA的批準(zhǔn)該藥是基于HELIOS-A 3的隨機(jī)��、開放標(biāo)簽��、全球性3期臨床試驗結(jié)果��。試驗結(jié)果顯示��,Amvuttra達(dá)到主要終點(diǎn),接受治療9個月后��,Amvuttra組的mNIS+7評分(評估神經(jīng)病障礙的評分)與基線相比降低2.2點(diǎn)(意味著癥狀改善)��,而外部對照組評分增加14.8點(diǎn)(意味著癥狀惡化��,p<0.0001)��。9個月時,50%的患者與基線相比癥狀得到改善��。Amvuttra同時達(dá)到試驗的所有次要終點(diǎn)��。并且在接受治療后18個月時��,療效數(shù)據(jù)與9個月時一致��。
17、Xenpozyme(Olipudase alfa)
2022年8月31日��,F(xiàn)DA批準(zhǔn)Genzyme公司的創(chuàng)新酶替代療法Olipudase alfa上市��,用于靜脈輸注治療酸性鞘磷脂酶缺乏癥(ASMD)的成人和兒童患者��。Olipudase alfa為首個用于ASMD患者非中樞神經(jīng)系統(tǒng)癥狀的藥物。ASMD由于缺乏分解復(fù)雜脂質(zhì)(稱為鞘磷脂)所需的酶引起的一種罕見遺傳病��,影響最嚴(yán)重的患者有嚴(yán)重的神經(jīng)系統(tǒng)癥狀��,其他患者也容易因為呼吸衰竭而過早死亡��。Olipudase alfa是一種酶替代療法��,有助于減少鞘磷脂在肝臟��、脾臟和肺中的積累��。
18��、Spevigo(Spesolimab)
2022年09月01日��,勃林格殷格翰宣布,美國FDA已批準(zhǔn)選擇性白細(xì)胞介素-36受體(IL-36R)抗體Spevigo(Spesolimab)上市��,用于治療泛發(fā)性膿皰性銀屑?�。℅PP)的發(fā)作��。Spevigo(Spesolimab)是FDA批準(zhǔn)的第1款GPP療法,這一批準(zhǔn)得到為期12周的關(guān)鍵性臨床試驗Effisayil 1的支持��。53名GPP發(fā)作患者接受了Spevigo或安慰劑的治療。接受治療一周后��,Spevigo組無可見膿皰的患者比例為54%��,安慰劑組這一數(shù)值為6%。在Effisayil™1中��,接受SPEVIGO的患者最常見的不良反應(yīng)(≥5%)是虛弱和疲勞��、惡心和嘔吐��、頭痛、瘙癢和瘙癢��、輸液部位血腫和瘀傷以及尿路感染��。
19��、Daxxify(daxibotulinumtoxinA)
2022年09月08日(田納西州納什維爾)Revance Therapeutics宣布,美國FDA已批準(zhǔn)Daxxify(DaxibotulinumtoxinA)上市��,用于暫時改善成人的中度至重度眉間紋。DAXXIFY™是第1個也是唯一1個通過肽交換技術(shù)™(PXT)穩(wěn)定的神經(jīng)調(diào)節(jié)劑��,不含人血清白蛋白和動物成分��。FDA的批準(zhǔn)是基于包含超過2700名參與者,接近4200次治療的3期臨床試驗項目的結(jié)果��。在關(guān)鍵性臨床試驗中��,74%的受試者在接受治療第4周��,依據(jù)研究者和患者的評估��,達(dá)到眉間紋改善兩級以上的水平��。中位療效持續(xù)時間為6個月��。最早療效可在接受治療1天后出現(xiàn)��。Daxxify表現(xiàn)出良好的安全性和耐受性,在臨床試驗中未報告嚴(yán)重治療相關(guān)不良事件��。
20��、ROLVEDON(eflapegrastim-xnst)
2022年09月09日��,Spectrum Pharmaceuticals,Inc.宣布��,美國FDA批準(zhǔn)ROLVEDON™(eflapegrastim-xnst)注射以減少感染的發(fā)生率(治療粒缺,同時作為粒缺伴發(fā)熱的支持藥物或者感染預(yù)防藥物)��。ROLVEDON™(eflapegrastim-xnst)注射液是20多年來首款獲FDA批準(zhǔn)的新型長效粒細(xì)胞集落刺激因子(G-CSF)��,通過結(jié)合在粒細(xì)胞祖細(xì)胞上表達(dá)的G-CSF受體來刺激其增殖過程��,使其最終在骨髓中產(chǎn)生功能性活化的嗜中性粒細(xì)胞,用于治療化療引起的中性粒細(xì)胞減少癥��。
ROLVEDON的BLA得到了來自兩項相同設(shè)計的3期��、隨機(jī)��、開放標(biāo)簽、非劣效性臨床試驗ADVANCE和RECOVER的數(shù)據(jù)的支持��,該試驗評估了ROLVEDON在643名早期乳腺癌患者中用于治療中性粒細(xì)胞減少癥的安全性和有效性由于骨髓抑制化療��。在這兩項研究中��,ROLVEDON在嚴(yán)重中性粒細(xì)胞減少癥(DSN)的平均持續(xù)時間中證明了預(yù)先指定的非劣效性(NI)假設(shè)以及與pegfilgrastim相似的安全性��。ROLVEDON在兩個試驗中的所有四個周期(所有NI p<0.0001)的平均DSN也證明了非劣于pegfilgrastim��。
21、Sotyktu(Deucravacitinib��,氘可來昔替尼)
2022年09月09日��,百時美施貴寶(BMS)宣布��,Sotyktu™(Deucravacitinib��,氘可來昔替尼)治療斑塊狀銀屑病的上市申請已獲FDA批準(zhǔn)��。Sotyktu是第一款獲FDA批準(zhǔn)的TYK2抑制劑��,F(xiàn)DA本次批準(zhǔn)是基于2項關(guān)鍵III期臨床試驗(POETYK PSO-1和POETYK PSO-2)的積極結(jié)果��。兩項試驗均為全球性��、多中心、雙盲��、隨機(jī)��、安慰劑和陽性藥物對照研究��,分別納入了666例患者和1020例患者,旨在評估氘可來昔替尼對比阿普米司特(apremilast)和安慰劑治療中重度斑塊狀銀屑病成人患者的療效和安全性��。共同的主要終點(diǎn)為第16周時銀屑病面積與嚴(yán)重性指數(shù)(PASI)評分改善75%以上和sPGA 0/1(靜態(tài)醫(yī)生總體評估皮膚癥狀完全清除/幾乎完全清除)的患者比例��。
數(shù)據(jù)顯示��,第16周時��,與安慰劑組和阿普米司特組相比,氘可來昔替尼組PASI評分改善75%以上和sPGA 0/1的患者比例顯著增加��。試驗還達(dá)到了所有次要終點(diǎn)��,氘可來昔替尼在癥狀負(fù)擔(dān)和生活質(zhì)量測量值上表現(xiàn)出顯著且具有臨床意義的改善��。此外��,氘可來昔替尼耐受性良好,因不良事件導(dǎo)致的停藥率低��。
22��、Terlivaz(Terlipressin)
2022年09月14日��,Mallinckrodt plc宣布美國食品藥品監(jiān)督管理局(FDA)批準(zhǔn)Terlivaz®(Terlipressin)特利加壓素注射劑��,改善肝腎綜合征(HRS)成人的腎功能迅速下降。Terlivaz是第1個也是唯一1個獲得FDA批準(zhǔn)的產(chǎn)品��,用于改善肝腎綜合征(Hepatorenal Syndrome��,HRS)成人的腎功能��,腎功能迅速下降��,HRS是一種需要住院治療的急性和危及生命的疾病��。FDA的批準(zhǔn)部分基于3期CONFIRM試驗的結(jié)果��,該試驗是有史以來規(guī)模最大的前瞻性研究(n=300)��,旨在評估特利加壓素在HRS 1型(HRS-1)患者中的安全性和有效性��。美國和加拿大。
CONFIRM試驗達(dá)到了驗證HRS逆轉(zhuǎn)的主要終點(diǎn)��,定義為腎功能改善��、避免透析和短期存活(p=0.012)��。1為實(shí)現(xiàn)經(jīng)驗證的HRS逆轉(zhuǎn),患者必須在第14天或出院前連續(xù)兩次血清肌酐(SCr)值≤1.5 mg/dL��,至少相隔兩個小時��。為納入主要療效終點(diǎn)分析��,患者必須在達(dá)到經(jīng)驗證的HRS逆轉(zhuǎn)后至少10天存活且未進(jìn)行腎臟替代治療(例如透析)��。初步結(jié)果在AASLD年會The Liver Meeting®2019的最新會議上公布��。結(jié)果還于2021年3月發(fā)表在《新英格蘭醫(yī)學(xué)雜志》上。CONFIRM試驗是在2021年AASLD肝腎綜合征指南中發(fā)布的更新診斷標(biāo)準(zhǔn)和術(shù)語之前完成的��。
與安慰劑相比��,在至少4%接受Terlivaz治療的患者中��,最常見的不良反應(yīng)是19.5%(n=39)的患者報告的腹痛(vs.6.1%;n=6),報告的惡心率為16%(n=32)患者(vs.10.1%;n=10)��,15.5%(n=31)患者報告呼吸衰竭(vs.7.1%;n=7)13%(n=26)報告腹瀉的患者(與7.1%��;n=7)和12.5%(n=25)的患者(與5.1%;n=5)報告了呼吸困難��。
23��、Elucirem(Gadopiclenol)
2022年9月21日��,F(xiàn)DA經(jīng)過優(yōu)先審查批準(zhǔn)了Guerbet公司的Elucireem™��,這是一種用于對比增強(qiáng)磁共振成像(MRI)的新型大環(huán)釓造影劑。該產(chǎn)品用于檢測和可視化中樞神經(jīng)系統(tǒng)(大腦��、脊柱和相關(guān)組織)和身體(頭頸部��、胸部��、腹部、骨盆和肌肉骨骼系統(tǒng))中的血管異常病變��。Gadopiclenol是Eucirem™的活性物質(zhì)��,有兩個水分子交換位點(diǎn)��,用以增加弛緩性和對比度��,與其他非特異性GBCA(大環(huán)釓基對比劑)相比,使用Gadopiclenol的劑量只有常規(guī)釓劑量的一半��,緩解了從業(yè)者對放射性暴露的擔(dān)憂��。
24��、Omlonti(Omidenepag Isopropyl)
2022年9月22日��,參天公司(Santen Inc.)和UBE Corporation(UBE)宣布,美國FDA批準(zhǔn)OMLONTI®(omidenepag isopropyl ophthalmic solution)0.002%奧米替尼異丙基滴眼液用于降低原發(fā)性開角型青光眼或高眼壓患者的眼壓升高(IOP)��。OMLONTI®在三項隨機(jī)對照臨床試驗中對開角型青光眼或高眼壓患者進(jìn)行了評估��,平均基線IOP為24-26 mm Hg��。
在所有三項研究中,雙盲治療持續(xù)時間均為三個月��。第三項研究包括在3個月的雙盲治療期之后的9個月的開放標(biāo)簽治療期��。在三項研究中��,觀察到所有治療組的眼壓降低。在OMLOTI®臂中��,所有三項研究的眼壓降低范圍為5-7 mm Hg��。噻嗎洛爾和拉坦前列素組的相應(yīng)降低分別為5-7 mm Hg和6-8 mm Hg��。
25��、Relyvrio(sodium phenylbutyrate+taurursodiol,苯丁酸+?�;切苋パ跄懰?
2022年09月29日��,美國FDA宣布��,批準(zhǔn)了Amylyx制藥公司開發(fā)的Relyvrio(sodium phenylbutyrate/taurursodiol)苯丁酸鈉和牛磺酸二醇口服固定劑量配方��,用于治療肌萎縮側(cè)索硬化(ALS)��。此次獲批是基于一項入組了137例ALS患者的2期臨床試驗獲得的積極數(shù)據(jù)��。試驗達(dá)到其主要療效終點(diǎn),即根據(jù)修訂的ALS功能評定量表測量��,在6個月隨機(jī)化階段結(jié)束時��,接受Relyvrio治療的ALS患者運(yùn)動功能下降顯著減緩。
26��、Lytgobi(Futibatinib)
2022年10月30日��,日本大鵬藥品(Taiho Pharmaceutical)和子公司Taiho Oncology宣布��,美國FDA已加速批準(zhǔn)Lytgobi(Futibatinib)片劑上市,用于治療攜帶FGFR2基因融合或其它重排的不可切除��、局部晚期或轉(zhuǎn)移性肝內(nèi)膽管癌經(jīng)治成人患者��。LYTGOBI的批準(zhǔn)是基于FOENIX*-CCA2試驗的初步分析結(jié)果��,這是一項全球2期開放標(biāo)簽試驗,評估了103名具有FGFR2基因重排(包括融合)的不可切除��、局部晚期或轉(zhuǎn)移性iCCA患者��。在這項試驗中��,患者每天口服一次LYTGOBI,劑量為20mg��,直至疾病進(jìn)展或出現(xiàn)不可接受的毒性��。試驗結(jié)果顯示��,futibatinib達(dá)到42%的客觀緩解率��,中位緩解持續(xù)時間為9.7個月��,72%的患者緩解持續(xù)時間超過6個月��。
27、Imjudo(Tremelimumab)
2022年10月21日��,美國FDA官網(wǎng)顯示��,阿斯利康(AstraZeneca)公司開發(fā)的抗CTLA-4抗體Imjudo(tremelimumab)已經(jīng)獲得批準(zhǔn)��,與抗PD-L1抗體Imfinzi(durvalumab)聯(lián)用��,治療不可切除的肝細(xì)胞癌患者��,為肝癌患者提供了一種由雙重免疫檢查點(diǎn)抑制劑構(gòu)成的全新免疫組合療法。
Tremelimumab和Imfinzi組成的聯(lián)合用藥方案(阿斯利康將其稱為STRIDE方案)對于肝細(xì)胞癌的療效和安全性得到了臨床研究積極結(jié)果的支持��,在一項針對不可切除的肝細(xì)胞癌患者的3期臨床試驗中��,研究者首先對患者進(jìn)行一次tremelimumab和Imfinzi的聯(lián)合用藥��,然后每隔4周進(jìn)行Imfinzi的單藥治療��,這一給藥方案旨在刺激T細(xì)胞激活的同時,減少CTLA-4抗體的毒副作用��。該試驗結(jié)果顯示��,接受這一聯(lián)合用藥方案治療的患者��,與活性對照組相比死亡風(fēng)險降低了22%(HR=0.78,96.02%CI:0.65-0.93��,p=0.0035)��,并且總生存期��、客觀緩解率、3年后患者的存活率等多項指標(biāo)均顯著優(yōu)于對照組患者��。
28��、Tecvayli(Teclistamab)
2022年10月25日��,強(qiáng)生(Johnson&Johnson)集團(tuán)旗下楊森(Janssen)公司宣布,美國FDA已經(jīng)加速批準(zhǔn)同時靶向B細(xì)胞成熟抗原(BCMA)和CD3受體的雙特異性抗體Tecvayli(Teclistamab)上市��,用于治療復(fù)發(fā)/難治性多發(fā)性骨髓瘤(RRMM)成人患者��。Teclistamab同時靶向BCMA與T細(xì)胞表面CD3受體的雙特異性抗體��,作為皮下治療給藥��。這種現(xiàn)成(或即用)療法使用創(chuàng)新科學(xué)��,通過將CD3陽性T細(xì)胞募集到表達(dá)BCMA的骨髓瘤細(xì)胞附近��,激發(fā)T細(xì)胞殺傷腫瘤細(xì)胞��。它為患者提供了一款現(xiàn)貨型��、皮下注射的治療選擇��。
29��、ELAHERE(Mirvetuximab soravtansine)
2022年11月14日��,ImmunoGen Inc.宣布美國FDA加速批準(zhǔn)抗體偶聯(lián)藥物(ADC)ELAHERE™(mirvetuximab soravtansine-gynx)用于治療葉酸受體α(FRα)陽性、鉑耐藥上皮性卵巢癌��、輸卵管癌或原發(fā)性腹膜癌的成年患者��,這些患者之前接受過1到3種全身治療方案��。
ELAHERE是一款的抗體偶聯(lián)藥物(ADC),包含葉酸受體α結(jié)合抗體��、可裂解接頭和美登木素生物堿有效載荷DM4(the maytansinoid payload DM4)��,DM4是一種有效的微管蛋白抑制劑,旨在殺死靶向癌細(xì)胞��。卵巢癌患者對含鉑療法產(chǎn)生耐藥性是成功控制疾病的一個重大挑戰(zhàn)��。
FRα是葉酸受體家族的一員��,它以高親和力與葉酸結(jié)合��,導(dǎo)致它們被內(nèi)吞攝入細(xì)胞內(nèi)。之前的研究表明��,F(xiàn)Rα在76-89%的上皮卵巢癌和35-68%的三陰性乳腺癌中高度表達(dá)��。使FRα成為引人關(guān)注的藥物靶點(diǎn)��。而且��,F(xiàn)Rα介導(dǎo)的信號通路能夠影響腫瘤細(xì)胞的分裂和遷移,因此抑制FRα還可能產(chǎn)生一定程度的直接抗癌活性��。
30��、TZIELD(teplizumab)
2022年11月18日��,Provention Bio,Inc.生物制藥公司宣布美國食品藥品監(jiān)督管理局(FDA)批準(zhǔn)了TZIELD(teplizumab-mzwv)的生物制品許可申請(BLA)��,用于延緩8歲及以上患有2期1型糖尿?�。═1D)的成人和3期1型糖尿?�。═1D)兒童患者的發(fā)作��。該療法的獲批是基于一項隨機(jī)、雙盲��、設(shè)置對照的臨床試驗��。有76名1型糖尿病患者參與了該試驗��,并被隨機(jī)分為兩組,一組接受teplizumab治療��,另一組則接受安慰劑��。治療一共維持了14天��,而在隨后的隨訪中��,治療組的44名患者里,有45%最后出現(xiàn)1型糖尿病的進(jìn)展,而對照組的數(shù)字則高達(dá)72%��。此外��,治療組患者出現(xiàn)疾病進(jìn)展的時間約為50個月��,而對照組的數(shù)字僅為25個月。這些數(shù)據(jù)表明teplizumab的治療可以顯著延緩1型糖尿病的病情進(jìn)展��。
31��、Rezlidhia(Olutasidenib)
2022年12月01日,Rigel Pharmaceuticals公司宣布��,美國FDA已經(jīng)批準(zhǔn)異檸檬酸脫氫酶1(IDH1)選擇性抑制劑Rezlidhia(olutasidenib)上市��,用于治療復(fù)發(fā)或難治性(R/R)急性髓系白血?�。ˋML)患者,這些患者具有易感異檸檬酸脫氫酶-1(IDH1)突變��。
Rezlidhia的的批準(zhǔn)基于針對147名患有復(fù)發(fā)性或難治性AML患者(都有IDH1突變)的臨床研究。療效的評估基于完全緩解率(CR)加上完全緩解伴部分血液學(xué)恢復(fù)(CRh)的比率、CR+CRh的持續(xù)時間��。CR+CRh率為35%��,CR+CRh的中位時間為1.9個月��,CR+CRh的中位持續(xù)時間為25.9個月��。
32��、Krazati(adagrasib)
2022年12月12日��,美國FDA宣布加速批準(zhǔn)Mirati公司KRAS G12C抑制劑Krazati(adagrasib)上市��,用于治療攜帶KRAS G12C突變的局部晚期或轉(zhuǎn)移性非小細(xì)胞肺癌(NSCLC)患者��,這些患者先前至少接受過一種全身性療法��。這是FDA批準(zhǔn)的第二款直接抑制KRAS突變體活性的靶向療法��。
FDA的批準(zhǔn)是基于adagrasib在支持注冊的2期臨床試驗KRYSTAL-1中的隊列結(jié)果��。在攜帶KRAS G12C突變的晚期NSCLC患者中��,adagrasib達(dá)到43%的客觀緩解率(ORR)和80%的疾病控制率(DCR)��。
33��、Lunsumio(Mosunetuzumab)
2022年12月22日��,羅氏(Roche)旗下基因泰克(Genentech)宣布��,美國FDA已批準(zhǔn)Lunsumio(Mosunetuzumab-axgb)用于治療經(jīng)過兩種或多種前期系統(tǒng)治療后復(fù)發(fā)或難治的濾泡性淋巴瘤(FL)成年患者��。此次FDA的批準(zhǔn)是基于2期GO29781研究的積極結(jié)果��,該試驗結(jié)果顯示出Lunsumio對于復(fù)發(fā)或難治濾泡性淋巴瘤患者具有較高且持久的應(yīng)答率��。在接受Lunsumio治療的患者中��,80%的患者出現(xiàn)了客觀反應(yīng)(包括完全反應(yīng)和部分反應(yīng))��,大多數(shù)患者至少18個月內(nèi)保持了反應(yīng)(57%)��。應(yīng)答者的中位應(yīng)答持續(xù)時間接近2年��。60%的患者獲得完全反應(yīng)(CR)��。
34��、Sunlenca(Lenacapavir)
2022年12月22日��,吉利德科學(xué)(Gilead Sciences)宣布FDA批準(zhǔn)其藥品Sunlenca(Lenacapavir)注射液和片劑的上市許可,用于聯(lián)合其他抗逆轉(zhuǎn)錄病毒藥物治療多重耐藥人類免疫缺陷病毒(HIV)感染的成人患者��。Sunlenca是首個基于衣殼抑制劑的HIV治療選項��,該藥的上市批準(zhǔn)主要基于2/3期臨床試驗CAPELLA數(shù)據(jù)的支持��,該試驗評估了Sunlenca聯(lián)合優(yōu)化背景抗病毒療法治療方案在接受過大量治療的多重耐藥HIV患者中的應(yīng)用��。
在顯著未滿足醫(yī)療需求的患者人群中��,83%(n=30/36)接受優(yōu)化背景方案的基礎(chǔ)上加用Sunlenca的受試者在第52周時達(dá)到病毒載量檢測不到的標(biāo)準(zhǔn)(<50拷貝/毫升)��。此外��,CAPELLA受試者的CD4陽性細(xì)胞計數(shù)平均增加82個細(xì)胞/微升��?�!缎掠⒏裉m醫(yī)學(xué)雜志》于今年5月發(fā)表了CAPELLA研究的主要結(jié)果��。
35、Xenoview(Hyperpolarized Xe-129)
2022年12月28日,F(xiàn)DA批準(zhǔn)Polarean Imaging開發(fā)的Xenoview(Hyperpolarized Xe-129)上市��,該藥是一種用于磁共振成像(MRI)的超極化造影劑,用于評估成人和12歲及以上兒童患者的肺通氣量��。當(dāng)吸入超極化氙Xe 129氣體時��,可以使用具有多核能力的MRI掃描儀對整個通氣肺中的超極化氙Xe 129分布進(jìn)行成像��。
36、NexoBrid(Anacaulase)
2022年12月28日��,MediWound公司宣布美國FDA已批準(zhǔn)其新藥NexoBrid(anacaulase-bcdb)上市��,用于移除有深度部分皮層燒傷和/或全皮層燒傷的成人患者身上的焦痂��。該藥物的上市申請基于一系列臨床前研究��,以及8項臨床試驗的數(shù)據(jù),其中包括一項關(guān)鍵的美國3期臨床試驗��。研究表明與對照相比��,該藥物可以達(dá)到95%及以上的焦痂移除��,達(dá)到主要臨床終點(diǎn)��。
此外,該療法也達(dá)到了一系列次要臨床終點(diǎn)��,包括焦痂移除時間更短、需要手術(shù)移除焦痂的比例更低��,以及與手術(shù)和非手術(shù)的標(biāo)準(zhǔn)護(hù)理方式相比��,失血更少等��。在安全性上��,與標(biāo)準(zhǔn)護(hù)理方式相比��,該療法也達(dá)到了非劣效性的臨床終點(diǎn)?�?傮w上看,該療法安全且耐受良好��。
37、BRIUMVI(Ublituximab)
2022年12月28日,TG Therapeutics,Inc.宣布��,美國FDA已批準(zhǔn)BRIUMVI™(ublituximab-xiiy)用于治療復(fù)發(fā)性多發(fā)性硬化癥(RMS)的各種形式,包括成人的臨床孤立綜合征��、復(fù)發(fā)緩解型疾病和活動性繼發(fā)性進(jìn)展性疾病��。Briumvi的獲批基于ULTIMATE I和ULTIMATE II兩項為期96周的3期試驗研究��,試驗在1,094名復(fù)發(fā)型MS患者中進(jìn)行��。接受Briumvi治療的患者的年化復(fù)發(fā)率(主要終點(diǎn))分別為0.076和0.091��,而特立氟

