基于全球出現(xiàn)的一系列創(chuàng)新型生物制品注冊(cè)分類與產(chǎn)品實(shí)際用途不匹配的問題,如預(yù)防用單克隆抗體和治療用疫苗��,本文對(duì)國內(nèi)外生物制品的分類和注冊(cè)申請(qǐng)路徑進(jìn)行比較�����,分析如何與時(shí)俱進(jìn)地調(diào)整注冊(cè)分類方法��,為創(chuàng)新生物制品的研發(fā)注冊(cè)提供適宜的監(jiān)管路徑和程序�����。
《生物制品注冊(cè)分類及申報(bào)資料要求》(2020年第43號(hào))規(guī)定�����,生物制品是指以微生物�����、細(xì)胞���、動(dòng)物或人源組織和體液等為起始原材料�����,用生物學(xué)技術(shù)制成�����,用于預(yù)防�����、治療和診斷人類疾病的制劑���。隨著科學(xué)技術(shù)不斷進(jìn)步���,生物制品更新迭代迅速,生物制品治療用和預(yù)防用的功能逐漸交疊���,單一類別的生物制品已經(jīng)無法用治療或者預(yù)防的功能進(jìn)行固化界定,全球范圍出現(xiàn)一系列創(chuàng)新型生物制品���,如預(yù)防用單克隆抗體(以下簡稱為單抗)和治療用疫苗等���。隨著新型生物制品的出現(xiàn),無法與現(xiàn)行生物制品注冊(cè)分類進(jìn)行匹配��。本文對(duì)國內(nèi)外生物制品的分類和注冊(cè)申請(qǐng)路徑進(jìn)行比較,分析如何與時(shí)俱進(jìn)調(diào)整注冊(cè)分類方法���,為創(chuàng)新生物制品的研發(fā)注冊(cè)提供適宜的監(jiān)管路徑和程序��。
1���、我國生物制品定義和注冊(cè)分類情況
我國生物制品注冊(cè)分類大體上分為2個(gè)階段,第1階段按產(chǎn)品列舉方式分類�����,第2階段按生物制品預(yù)期用途進(jìn)行分類�����。
1.1生物制品注冊(cè)按產(chǎn)品列舉式分類
1951年頒布的《生物制品法規(guī)》是我國最早的一部關(guān)于生物制品的法規(guī)���,含12個(gè)通則和26個(gè)品種各論��,是最早與疫苗有關(guān)的質(zhì)量標(biāo)準(zhǔn)�����。1985年之前��,我國的生物制藥產(chǎn)業(yè)仍以傳統(tǒng)生物制品為主���,即疫苗和血液制品��,總體上這個(gè)階段批準(zhǔn)的生物制品還是(預(yù)防用)疫苗占主導(dǎo)地位���。
1985年衛(wèi)生部頒布了第一部《新生物制品審批辦法》,首次將新生物制品按照產(chǎn)品類別劃分為4項(xiàng)注冊(cè)分類���。1988年頒布《新生物制品審批辦法補(bǔ)充規(guī)定》���,新生物制品與新藥品難以區(qū)分者按以下辦法處理。原則上按其用途���,用作免疫預(yù)防者屬于生物制品�����;用作臨床治療者屬于藥品;如該產(chǎn)品的生產(chǎn)工藝���、質(zhì)量控制標(biāo)準(zhǔn)及其檢測方法接近生物制品者�����,雖屬治療用品仍按新生物制品審批程序申報(bào)���,申報(bào)時(shí)需參考《新藥審批辦法》中有關(guān)技術(shù)要求�����。1993年原衛(wèi)生部頒布《生物制品管理規(guī)定》�����,仍沿用產(chǎn)品列舉式分類���。
1.2生物制品注冊(cè)按預(yù)期用途分類
隨著生物制品種類增多,按產(chǎn)品列舉式分類變得愈發(fā)困難�����,1999年新修訂的《新生物制品審批辦法》中將生物制品申報(bào)按照用途分為治療用新生物制品�����、預(yù)防用新生物制品和體外診斷用品。
2002年��,國家藥品監(jiān)督管理局頒布《藥品注冊(cè)管理辦法(試行)》(局令第35號(hào)) �����,細(xì)化了生物制品的注冊(cè)分類�����,將治療用生物制品分為15類���,包括單抗��、基因治療���、體細(xì)胞治療及其制品等。預(yù)防用生物制品也分為15類�����,均為疫苗��。2005年��,原國家食品藥品監(jiān)督管理局頒布《藥品注冊(cè)管理辦法》以及2007年修訂均延續(xù)了上述分類方式�����。
《藥品注冊(cè)管理辦法》2020年版自2020年7月1日正式實(shí)施��。生物制品注冊(cè)按照生物制品創(chuàng)新藥���、生物制品改良型新藥�����、已上市生物制品(含生物類似藥) 等進(jìn)行分類���。為規(guī)范生物制品注冊(cè)申報(bào)和管理,《生物制品注冊(cè)分類及申報(bào)資料要求》(2020年6月)將生物制品分為預(yù)防用生物制品���、治療用生物制品和按生物制品管理的體外診斷試劑��。其中�����,預(yù)防用生物制品是指為預(yù)防��、控制疾病的發(fā)生���、流行���,用于人體免疫接種的疫苗類生物制品,包括免疫規(guī)劃疫苗和非免疫規(guī)劃疫苗�����,見表1���。
2���、新型生物制品帶來的注冊(cè)分類問題和挑戰(zhàn)
隨著生物制藥技術(shù)的飛速發(fā)展,越來越多的新型生物制品申報(bào)上市���,一方面�����,以單抗為代表的非疫苗類預(yù)防用生物制品出現(xiàn)��;另一方面�����,同一類生物制品可能同時(shí)兼有預(yù)防用和治療用等多重功能���。目前生物制品的分類方式,將預(yù)防用和治療用功能截然分開��,且預(yù)防用生物制品等同為疫苗可能導(dǎo)致新型生物制品的注冊(cè)分類難題�����。
2.1新型長效單抗無法納入預(yù)防用生物制品類別
單抗是由單一雜交瘤細(xì)胞產(chǎn)生���,針對(duì)單一抗原表位的特異性抗體���。自1986年首個(gè)鼠源單抗muromonab-CD3經(jīng)美國FDA批準(zhǔn)上市用于治療器官移植后同種異體排斥反應(yīng)至今,全球已有100多種單抗獲批上市���,單抗在癌癥�����、自身免疫性疾病和感染性疾病等疾病中發(fā)揮重要作用��。
長期以來�����,單抗不可避免的高研發(fā)成本所致的昂貴售價(jià)并不親民��,并且頻繁地注射給藥也給患者帶來諸多不便��。因此�����,近年來���,延長單抗半衰期成為抗體工程改造的重點(diǎn)�����。單抗半衰期的延長不僅可以減少給藥頻率��,降低治療費(fèi)用���,并且使單抗藥物用于感染性疾病的預(yù)防變?yōu)榭赡堋慰乖瓉戆胨テ诩s為11~30d�����,經(jīng)抗體工程改造后的單抗半衰期長達(dá)2~4個(gè)月。目前的生物制品分類方式��,預(yù)防用生物制品等同為疫苗可能導(dǎo)致全球范圍內(nèi)出現(xiàn)的創(chuàng)新型預(yù)防用長效單抗無法納入預(yù)防用生物制品類別��。
2.1.1預(yù)防呼吸道合胞病毒(respiratory syncytial virus�����,RSV)感染長效單抗
RSV是全球5歲以下兒童急性下呼吸道感染最常見的病毒病原��,也是導(dǎo)致1歲以下嬰兒罹患下呼吸道感染住院的首要病毒病原���。目前,尚無RSV疫苗和有效的抗病毒藥物用于RSV感染的防治�����。一種可選擇的預(yù)防RSV的方法是用單抗進(jìn)行被動(dòng)免疫預(yù)防��。帕利珠單抗是目前唯一被FDA批準(zhǔn)用于RSV被動(dòng)免疫預(yù)防的單抗���,僅用于患有先天性心臟病或肺部疾病的不足35周的早產(chǎn)兒��,且半衰期短���,需要每周注射�����。長效��、全人源預(yù)防用RSV單抗一直是全球各國研發(fā)的熱點(diǎn)���。預(yù)防用長效單抗nirsevimab是首個(gè)完成全球Ⅲ期臨床試驗(yàn)的單抗,與帕利珠單抗相比中和RSV活性高50倍��,具有超長半衰期�����,并擴(kuò)大適用人群為所有嬰幼兒�����。其中�����,Ⅲ期臨床研究證明單次肌內(nèi)注射nirsevimab可在整個(gè)RSV流行季,給晚期健康早產(chǎn)兒及足月兒提供持續(xù)5個(gè)月的保護(hù)��。
目前�����,我國已有3種用于預(yù)防RSV引起的下呼吸道疾病的長效單抗獲批進(jìn)入臨床試驗(yàn)階段��,目標(biāo)人群包括0~2歲的健康嬰幼兒���、早產(chǎn)兒和具有RSV高危因素的嬰幼兒�����。盡管3款種長效單抗均通過被動(dòng)免疫用于疾病預(yù)防,但由于不是疫苗�����,不符合我國目前預(yù)防用生物制品(等同為疫苗)的注冊(cè)分類���,無法納入預(yù)防用生物制品類別�����,因此在臨床試驗(yàn)受理階段暫納入治療用生物制品注冊(cè)分類��,見表2��。

2.1.2預(yù)防瘧疾感染長效單抗
瘧疾是瘧原蟲感染所致的地方性傳染病��,主要流行于熱帶和亞熱帶地區(qū)���,疾病病死率較高��。根據(jù)世界衛(wèi)生組織最新報(bào)告��,2019年全球報(bào)告2.29億例瘧疾病例��,逾40.9萬例死于瘧疾���。目前已有3個(gè)用于預(yù)防(和治療)瘧疾感染的單抗進(jìn)入臨床試驗(yàn)階段(見表3),其中長效單抗L9LS是CIS43LS的下一代單抗���,其針對(duì)的是CSP-1蛋白的一個(gè)更緊密的區(qū)域�����,具有高親和力���,保護(hù)效力是CIS43LS的3倍���。meplazumab是靶向CD147的單抗藥物,2020年1月獲美國FDA批準(zhǔn)開展用于治療和預(yù)防嚴(yán)重瘧疾的Ⅰ期臨床試驗(yàn)��。

2022年8月4號(hào)�����,一項(xiàng)旨在預(yù)防瘧疾感染的新型單抗L9LS的Ⅰ期臨床試驗(yàn)結(jié)果在《新英格蘭醫(yī)學(xué)雜志》上發(fā)表���。試驗(yàn)旨在評(píng)估L9LS對(duì)從未患過瘧疾或未接種瘧疾疫苗的健康成年人的安全性和藥動(dòng)學(xué)���,以及人瘧疾感染的保護(hù)效力���。試驗(yàn)通過皮下注射或靜脈注射給予L9LS��,在接受抗體注射的2~6周內(nèi)��,讓所有參與者都暴露于攜帶瘧原蟲的蚊子��,暴露后幾周內(nèi)�����,研究人員觀察到幾乎所有的受試者都得到了保護(hù)��。L9LS經(jīng)抗體工程優(yōu)化(LS突變)��,半衰期為56d���,對(duì)于5歲以下的兒童���,1次皮下注射可能提供至少6個(gè)月的保護(hù)效力。
2.1.3新型冠狀病毒肺炎(COVID-19)暴露前預(yù)防長效單抗
COVID-19暴發(fā)以來�����,疫苗在控制病毒傳播和降低死亡率方面發(fā)揮著重要作用��,是向大多數(shù)人提供COVID-19保護(hù)的最適當(dāng)措施�����。此外���,在未接種疫苗或?qū)σ呙缬胁涣挤磻?yīng)的人群(如免疫功能低下的個(gè)人)中���,長效單抗可提供暴露前被動(dòng)免疫預(yù)防作用�����。
恩適得(Evusheld��,tixagevimab與cilgavimab的組合)是目前全球唯一一款用于COVID-19暴露前預(yù)防的單抗組合藥物���,2021年12月獲得美國FDA的緊急使用授權(quán),2022年3月獲得歐洲EMA上市許可�����,2022年6月通過海南博鰲樂城國際醫(yī)療旅游先行區(qū)特殊進(jìn)口審批�����,適用于成人和青少年(年齡≥12歲且體重≥40 kg)的新冠病毒暴露前預(yù)防��。
Evusheld獲批美國FDA緊急使用授權(quán)是基于PROVENTⅢ期暴露前預(yù)防臨床試驗(yàn)數(shù)據(jù)��,與安慰劑相比�����,Evusheld組受試者出現(xiàn)COVID-19癥狀的風(fēng)險(xiǎn)在統(tǒng)計(jì)學(xué)上顯著降低(初步分析為77%���,中位6個(gè)月隨訪時(shí)為83%)�����,病毒防護(hù)效果持續(xù)至少6個(gè)月��。
2.2同類產(chǎn)品同時(shí)具有預(yù)防和治療功能
隨著免疫學(xué)研究的發(fā)展,疫苗的研發(fā)和應(yīng)用逐漸延伸���,從傳染病擴(kuò)大到系統(tǒng)性疾病,包括癌癥���;從預(yù)防用擴(kuò)大到治療用�����。
全球范圍內(nèi)�����,卡介苗主要用作預(yù)防兒童結(jié)核病的預(yù)防接種疫苗��,接種對(duì)象為出生3個(gè)月以內(nèi)的嬰兒或用舊結(jié)核菌素試驗(yàn)陰性的兒童���。除用作結(jié)核病疫苗外�����,卡介苗還是非肌層浸潤性膀胱癌的主要治療手段�����。1976年��,Morales醫(yī)生首次將卡介苗直接注入膀胱���,治療復(fù)發(fā)性淺表膀胱癌獲得成功。隨后大量的臨床觀察和研究表明���,卡介苗灌注治療術(shù)后殘存膀胱癌的完全緩解率為50%~90%(平均70%)��,有效降低了膀胱癌復(fù)發(fā)率��,推遲癌癥復(fù)發(fā)和病情進(jìn)展���。盡管卡介苗預(yù)防結(jié)核病和治療膀胱癌的用法不同��,但都是通過激活機(jī)體的免疫系統(tǒng),增強(qiáng)主動(dòng)免疫功能發(fā)揮藥理作用�����。
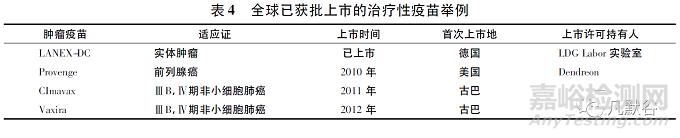
除卡介苗之外��,全球已有多款治療性疫苗上市��,見表4�����。2010年4月29日���,美國FDA批準(zhǔn)Provenge疫苗���,是以患者自身免疫細(xì)胞制成的前列腺癌免疫療法,開創(chuàng)了癌癥免疫治療的新時(shí)代�����。疫苗可以是治療用或者預(yù)防用��,但是本質(zhì)上是同一類通過主動(dòng)免疫產(chǎn)生免疫應(yīng)答的產(chǎn)品。而目前的生物制品分類方式�����,將產(chǎn)品的預(yù)防用和治療用功能截然分開�����。
2.3基于免疫學(xué)原理看生物制品的創(chuàng)新問題
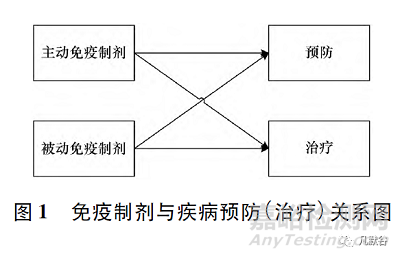
人工免疫是指人為地使機(jī)體獲得適應(yīng)性免疫���,包括2種:人工主動(dòng)免疫是用疫苗接種機(jī)體���,使之主動(dòng)產(chǎn)生適應(yīng)性免疫應(yīng)答,從而預(yù)防和治療疾病的措施��;人工被動(dòng)免疫是給人體注射含特異性抗體等制劑�����,使之被動(dòng)獲得適應(yīng)性免疫應(yīng)答���,以治療或緊急預(yù)防疾病的措施��,見圖1�����。目前生物制品的注冊(cè)分類方式���,主動(dòng)免疫制劑納入預(yù)防用生物制品,而被動(dòng)免疫制劑納入治療用生物制品�����。
2.3.1被動(dòng)免疫制劑和主動(dòng)免疫制劑都可以用于預(yù)防疾病
一個(gè)世紀(jì)以來�����,被動(dòng)免疫制劑已成功應(yīng)用于狂犬病���、破傷風(fēng)和乙型肝炎等一系列疾病疑似暴露的暴露后預(yù)防或感染后的輔助治療��。隨著科學(xué)技術(shù)飛速發(fā)展���,以健康人預(yù)防用長效單抗為代表的被動(dòng)免疫制劑可能為某些傳染病的預(yù)防提供干預(yù)手段,如RSV��、瘧疾等。因?yàn)檫@些疾病疫苗研發(fā)難度大���,目前仍缺乏有效預(yù)防手段�����,造成嚴(yán)重的疾病負(fù)擔(dān)�����。世界衛(wèi)生組織為此推出了一系列單抗預(yù)防疾病的理想產(chǎn)品特性清單�����。
盡管被動(dòng)免疫保護(hù)效力較短�����,但用單抗進(jìn)行被動(dòng)免疫相較于疫苗進(jìn)行主動(dòng)免疫有以下優(yōu)勢:①被動(dòng)免疫起效快���,即時(shí)就會(huì)有保護(hù)作用。而且在免疫抑制的個(gè)體中仍具有保護(hù)效力���,這些個(gè)體往往疾病感染風(fēng)險(xiǎn)最高��。②新技術(shù)可以簡化單抗的生產(chǎn)或給藥���,如延長抗體的半衰期或注射編碼單抗的mRNA�����。③生產(chǎn)過程通用��,原則上可以在短時(shí)間快速開始生產(chǎn)���,在疫情暴發(fā)時(shí)迅速提供產(chǎn)品���。
2.3.2被動(dòng)免疫制劑和主動(dòng)免疫制劑都可以用于治療疾病
免疫治療是指利用免疫學(xué)原理���,針對(duì)疾病的發(fā)生機(jī)制,人為地干預(yù)或調(diào)整機(jī)體的免疫功能��,達(dá)到治療疾病目的所采取的措施���,包括主動(dòng)或被動(dòng)免疫治療���。
美國批準(zhǔn)抗前列腺癌治療性疫苗上市���,促進(jìn)了抗腫瘤治療性疫苗的研發(fā),治療性疫苗已然成為醫(yī)藥生物技術(shù)與產(chǎn)業(yè)化的熱點(diǎn)之一��。治療性疫苗是指機(jī)體在感染或發(fā)生疾病后�����,用誘導(dǎo)機(jī)體產(chǎn)生特異性免疫或非特異性免疫的方法�����,防止疾病的發(fā)生��、發(fā)展��,或促進(jìn)已產(chǎn)生疾病的機(jī)體恢復(fù)健康�����,多用于慢性感染或腫瘤的治療���。各種治療性疫苗的作用機(jī)制與途徑不盡相同��,但最基本的理論基礎(chǔ)總是圍繞如何調(diào)節(jié)機(jī)體的免疫應(yīng)答�����,或增強(qiáng)或降低���,以達(dá)到對(duì)所患疾病有不同程度的緩解��、控制甚至治愈的效果�����。
基于免疫學(xué)原理��,預(yù)防疾病的生物制品除了主動(dòng)免疫制劑���,如疫苗類生物制品��,還包括以預(yù)防為目的的被動(dòng)免疫制劑��,如健康人預(yù)防用單抗等新型生物制品���。
3��、典型國家和地區(qū)生物制品分類和注冊(cè)申請(qǐng)路徑
3.1生物制品分類
3.1.1美國的生物制品分類
FDA對(duì)“生物制品”定義為:由FDA監(jiān)管���,用于診斷��、預(yù)防���、治療和治愈疾病和病癥,通常是一類大而復(fù)雜的分子���,并且種類繁多的產(chǎn)品���。此類產(chǎn)品通常運(yùn)用生物技術(shù)在生物體中產(chǎn)生,如微生物�����、植物細(xì)胞或動(dòng)物細(xì)胞�����,并且比小分子藥物更難表征�����。
2003年6月30日��,F(xiàn)DA將生物制品評(píng)價(jià)和研究中心(CBER)審評(píng)和監(jiān)管的一些生物制品轉(zhuǎn)移到藥品評(píng)價(jià)和研究中心(CDER)。CDER對(duì)轉(zhuǎn)移的生物制品負(fù)有監(jiān)管責(zé)任���,兩大中心共同促進(jìn)美國生物制品監(jiān)管的發(fā)展�����。其中細(xì)胞產(chǎn)品���,基因治療產(chǎn)品,疫苗和疫苗相關(guān)產(chǎn)品��,變態(tài)反應(yīng)原制品���,抗毒素���、抗蛇毒血清和蛇毒,血液��、血液成分���、血漿衍生品,人體細(xì)胞��、組織、細(xì)胞-組織產(chǎn)品歸屬CBER監(jiān)管��。單抗���、治療用蛋白(疫苗和血液制品除外)���、免疫調(diào)節(jié)劑、生長因子��、細(xì)胞因子歸屬于CDER監(jiān)管�����。
3.1.2歐盟的生物制品分類
在歐盟���,生物制品包括3類:使用生物技術(shù)生產(chǎn)的生物制品�����、先進(jìn)治療產(chǎn)品(ATMP)和其他生物制品���。使用生物技術(shù)生產(chǎn)的生物制品申請(qǐng)由EMA的人用藥品委員會(huì)(CHMP)負(fù)責(zé)審評(píng),先進(jìn)治療產(chǎn)品由先進(jìn)療法委員會(huì)(CAT)負(fù)責(zé)評(píng)估產(chǎn)品的質(zhì)量、有效性和安全性���,以上2類產(chǎn)品通過集中審批程序獲得授權(quán)�����。其他生物制品(如天然衍生生物制品)可以在各個(gè)成員國獲得國家授權(quán)�����,而不通過集中審批程序��。
3.1.3日本的生物制品分類
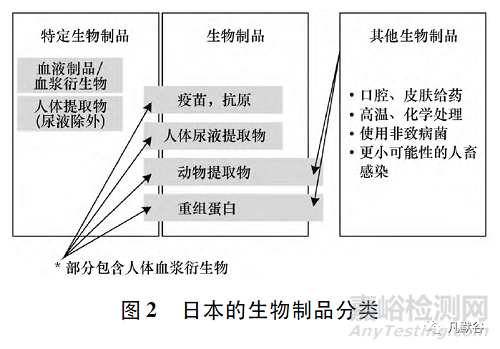
在日本���,對(duì)生物制品的潛在感染傳播風(fēng)險(xiǎn)進(jìn)行合理科學(xué)評(píng)估的基礎(chǔ)上將生物制品分為特定生物制品、生物制品和其他生物制44品(見圖2)�����。此外���,日本將細(xì)胞和基因產(chǎn)品等獨(dú)立出來歸于再生醫(yī)療產(chǎn)品�����,進(jìn)行單獨(dú)管理��,并在修訂的《藥品和醫(yī)療器械法》中增訂了再生醫(yī)學(xué)產(chǎn)品監(jiān)管的部分�����。
3.1.4生物制品的分類差異
除我國將生物制品分為治療用�����、預(yù)防用和診斷用進(jìn)行注冊(cè)分類外�����,美國���、歐盟和日本并未進(jìn)行這樣劃分,見表5���。國際上對(duì)生物制品分類的考慮因素包括生物制品的生產(chǎn)來源���、生物技術(shù)的先進(jìn)性、分子大小和表征復(fù)雜性等�����。歐盟和日本在傳統(tǒng)生物制品之外,分別增加先進(jìn)療法和再生醫(yī)學(xué)產(chǎn)品的分類���。

通常,生物制品注冊(cè)分類由各國藥品監(jiān)管機(jī)構(gòu)制定用于生物制品上市審評(píng)審批��,由于各國監(jiān)管機(jī)構(gòu)對(duì)生物制品的分類考量因素不同��,因此各國分類各異��。但是考慮到部分新型生物制品��,包括先進(jìn)療法藥品等的研發(fā)創(chuàng)新�����,具有區(qū)別于傳統(tǒng)藥物的獨(dú)特屬性�����,包括治愈潛力�����、一次性治療、高額的前期費(fèi)用��、復(fù)雜的生產(chǎn)過程和嚴(yán)格的運(yùn)輸條件��,因此在生物制品注冊(cè)分類時(shí)納入“先進(jìn)性”的考量因素��,有助于促進(jìn)創(chuàng)新��,提高ATMPs等新型生物制品的可及性���。
3.2注冊(cè)申請(qǐng)路徑
3.2.1美國的生物制品注冊(cè)申請(qǐng)路徑
①新生物制品注冊(cè)申請(qǐng)路徑。生物制品通過生物制品許可申請(qǐng)(BLA)路徑上市��,其中新生物制品一般通過PH-SA351(a)路徑申請(qǐng)��,在該路徑下需要完整的CMC���、非臨床以及臨床研究數(shù)據(jù)��,即包含完整研發(fā)資料的生物制品上市申請(qǐng)���。②生物類似藥注冊(cè)申請(qǐng)路徑。2010年修訂的PHSA中351(k)節(jié)中授予了FDA批準(zhǔn)生物類似藥的權(quán)利并且設(shè)立了生物類似藥簡化申請(qǐng)路徑���。生物類似藥只要證明與FDA已許可的生物制品具有生物相似性或可互換性即可通過簡化申請(qǐng)路徑申請(qǐng)���。
3.2.2歐盟的生物制品注冊(cè)申請(qǐng)路徑
歐盟對(duì)所有生物制品的注冊(cè)申請(qǐng)路徑分為2類:①完整資料申請(qǐng)路徑���,歐盟2001/83/EC指令第8條第3款規(guī)定,完整資料申請(qǐng)應(yīng)當(dāng)提交全面���、完整且沒有任何研究/試驗(yàn)報(bào)告的簡化�����,包括藥學(xué)(物理化學(xué)��、生物�����、微生物)試驗(yàn)�����、非臨床與臨床試驗(yàn)結(jié)果等在內(nèi)的申報(bào)資料���。②生物類似藥的簡化申請(qǐng)路徑��,生物類似藥申請(qǐng)��,即滿足2001/83/EC第十條第四款規(guī)定的簡化申請(qǐng)�����。簡化申請(qǐng)與完整申請(qǐng)相比,在非臨床研究以及臨床試驗(yàn)部分��,可以提交簡化的申報(bào)資料��,或者只需要提交生物利用度研究證明生物等效性即可��。
3.2.3日本的生物制品注冊(cè)申請(qǐng)路徑
日本的藥品審評(píng)注冊(cè)分類不劃分為“化學(xué)藥品”和“生物制品”��,而是以“新藥”�����、“改良型”和“仿制藥”(生物類似藥)的基本邏輯來劃分��。具體包括:①含有新有效成分的藥品�����。②新醫(yī)療用復(fù)方制劑。③新給藥途徑藥品���。④新適應(yīng)證藥品���。⑤新劑型藥品。⑥新用量藥品���。⑦生物類似物��。⑧增加新劑型的藥品���。⑨類似處方醫(yī)療用復(fù)方制劑。⑩其他處方藥��。
依據(jù)2005年3月31日�����,日本厚生勞動(dòng)省醫(yī)藥食品局發(fā)布《關(guān)于藥品生產(chǎn)銷售的批準(zhǔn)申請(qǐng)通知》(第0331015號(hào)令)�����。按照藥品的創(chuàng)新程度以及申報(bào)資料的完整程度���,注冊(cè)分類1類到10類的處方藥品�����,隨著其創(chuàng)新難度的降低���,其注冊(cè)申請(qǐng)所要求的資料也逐漸簡化���。
3.2.4生物制品的注冊(cè)申請(qǐng)路徑差異
從各國生物制品的注冊(cè)申請(qǐng)路徑來看�����,只有我國把生物制品分為治療用和預(yù)防用2類��。美國和歐盟的生物制品注冊(cè)分類按照“創(chuàng)新”和“仿制”(生物類似藥)的原則��,將注冊(cè)申請(qǐng)路徑分為完整申請(qǐng)路徑和簡化申請(qǐng)路徑��,日本按照藥品的創(chuàng)新程度以及申報(bào)資料的完整程度將注冊(cè)分類分成1~10類�����,見表6��。


4��、關(guān)于生物制品分類體系的思考與建議
4.1建立持續(xù)改進(jìn)的生物制品分類體系
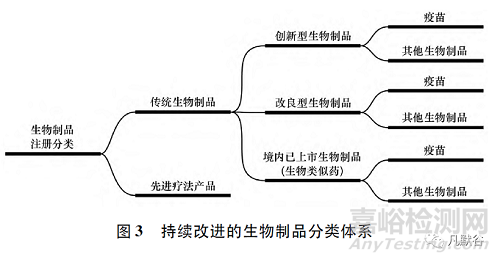
科學(xué)的本質(zhì)在于分類�����,對(duì)生物制品的注冊(cè)分類進(jìn)行適時(shí)調(diào)整是適應(yīng)生物制品創(chuàng)新和產(chǎn)業(yè)發(fā)展��,緊跟科學(xué)技術(shù)進(jìn)步的必然要求��。隨著科學(xué)和技術(shù)的發(fā)展���,生物制品治療與預(yù)防的功能逐漸交疊�����,未來生物制品的注冊(cè)分類不適合按照產(chǎn)品類別進(jìn)行預(yù)防用和治療用的簡單功能區(qū)分��,應(yīng)當(dāng)按照國際通行的創(chuàng)新��、改良和生物類似藥的分類邏輯進(jìn)行申報(bào)資料規(guī)定��?�?紤]到我國一直以來對(duì)疫苗有單獨(dú)注冊(cè)要求��,建議新的生物制品注冊(cè)分類見圖3��。
4.2區(qū)分傳統(tǒng)生物制品與先進(jìn)療法產(chǎn)品
先進(jìn)療法產(chǎn)品包括基因治療產(chǎn)品��、細(xì)胞治療產(chǎn)品和組織工程產(chǎn)品���,這些高度復(fù)雜的治療產(chǎn)品無論是生產(chǎn)和給藥途徑�����,還是臨床療效均不同于傳統(tǒng)藥物�����。一些基因治療產(chǎn)品可以解決疾病的病因,為患者提供一次性治愈的機(jī)會(huì)��,也有一些細(xì)胞治療產(chǎn)品和組織工程產(chǎn)品是專門為特定患者而生產(chǎn)的�����,形成個(gè)體化定制藥物�����。將先進(jìn)療法產(chǎn)品與傳統(tǒng)生物制品進(jìn)行注冊(cè)區(qū)分(圖3),有利于監(jiān)管部門制定一系列監(jiān)管策略�����,支持新的先進(jìn)療法產(chǎn)品的早期開發(fā)��,以便促進(jìn)患者可以更早獲得這類產(chǎn)品�����。
5���、結(jié)語
隨著生物制藥技術(shù)的飛速發(fā)展���,全球涌現(xiàn)出一系列的創(chuàng)新型生物制品,如預(yù)防用單抗和治療用疫苗等��。目前生物制品的注冊(cè)分類按照產(chǎn)品類別進(jìn)行預(yù)防用和治療用的簡單功能區(qū)分���,新型生物制品可能將面臨注冊(cè)分類與實(shí)際用途不匹配的問題���。建立持續(xù)改進(jìn)的生物制品分類體系��,區(qū)分傳統(tǒng)生物制品和先進(jìn)療法產(chǎn)品��,有利于生物制品的創(chuàng)新和產(chǎn)業(yè)發(fā)展�����,便于監(jiān)管部門制定監(jiān)管策略��,助力患者更早獲得創(chuàng)新產(chǎn)品��。