據(jù)統(tǒng)計(jì)����,在美國每個(gè)被批準(zhǔn)上市的新藥��,平均研發(fā)成本高達(dá)20億美元��,平均研發(fā)周期超過10年�。
而且新藥研發(fā)的成功率?常低�,有很多藥品從概念到成功上市,成功率只有5%�,可謂是「百?挑?」。每個(gè)成功上市的新藥背后�,都凝聚著眾多專家,投資?和企業(yè)的心血�。為了保證市場的健康成長,同時(shí)?勵(lì)創(chuàng)新和研發(fā)��,美國食品與藥品管理局(Food and Drug Administration, FDA)制定了?系列非常完善的監(jiān)管手段�。
在美國,?個(gè)普通的新化合物從最初的發(fā)現(xiàn)到申請上市��,大約需要經(jīng)過15年的時(shí)間����,其中 FDA 用于審評的時(shí)間大約為6到10個(gè)月�。美國的聯(lián)邦食品����、藥品、化妝品法案 FDCA(Federal Food, Drug and Cosmetic Act)規(guī)定了新藥審評的過程����,此法案有關(guān)新藥審評最重要的條款相當(dāng)簡單明了。
FDCA 規(guī)定����,任何?于診斷、治愈����、緩解、預(yù)防人����、其它動物疾病的物品,以及用于影響?����、或其它動物身體的結(jié)構(gòu)�、或功能的物品(?品除外)為藥品��。
FDCA 還規(guī)定��,任何新藥在上市之前?定要表明它是安全����、有效并且經(jīng)過審批的��。
新藥申請條件
符合以下任何情況均可以向 FDA 提出 NDA 申請:
1�、新分子實(shí)體(NME)
2、新化學(xué)實(shí)體(NCE)
3����、原批準(zhǔn)藥品相同化學(xué)成分的新鹽基、新酯基
4�、原批準(zhǔn)藥品的新配方組成
5、原批準(zhǔn)藥品的新適應(yīng)癥(包括處方藥轉(zhuǎn)非處方藥使用)
6�、新劑型、新給藥途徑��、新規(guī)格(單位含量)
7��、兩種以上原批準(zhǔn)藥品的新組合
新藥注冊途徑
505(b)指的是 FDCA 第5章第505分章�,即505法案��。
505法案包括NDA的3種申請:
1����、505b1:申報(bào)者進(jìn)?所有藥學(xué)研究(Completely new)
2����、505b2:同樣也是申報(bào)者進(jìn)?所有藥學(xué)研究,但不同的是部分信息不實(shí)由申報(bào)者??完成(by)��,或者這些研究不是為了申報(bào)者?完成的(for)����;以及申報(bào)者沒有引?的權(quán)利(Hybrid new, Some studyyoucan bridge)
3、505j:欲申報(bào)制劑在 API�,劑型,給藥途徑��,標(biāo)簽��,質(zhì)量�,檢驗(yàn),適應(yīng)癥上都和已有品種?樣�。(Generic, ANDA)
505j 是 ANDA 的注冊申請途徑,申請流程與 NDA 有所不同��,我們今后會詳細(xì)單獨(dú)描述,本?主要總結(jié)整理NDA 的注冊申請程序�。
(圖:新藥注冊途徑)
新藥申請程序
美國每年上市許多新藥,雖品種不同����,F(xiàn)DA 對它們的評審要求也各不相同,但評審框架是?致的����,大致可為以下幾個(gè)步驟:
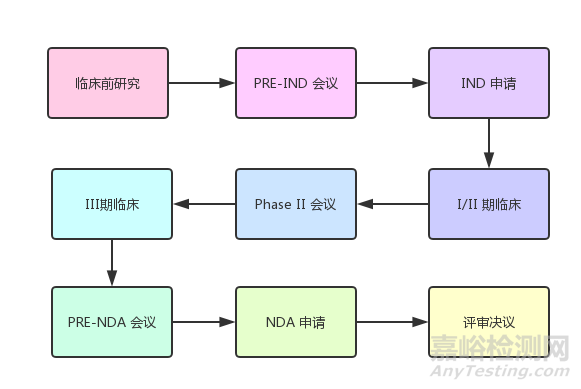
(圖:新藥申請程序)
1. 臨床前研究
新藥安全��、有效性的研究最終將在?體上進(jìn)?�,但在FDA允許試驗(yàn)藥物試?于人體之前,必須證明該藥的研究對?體是安全的��。如果新藥申請者不能從現(xiàn)有的研究數(shù)據(jù)����、本國及他國的使?等數(shù)據(jù)證明該藥是安全的,那么就必須要進(jìn)行臨床前研究�。
在這階段,F(xiàn)DA?般規(guī)定(最低限度)新藥申請者必須:
① 完成該藥的藥理研究
② 在?少?種動物身上進(jìn)行急毒試驗(yàn)
③ 按照該藥預(yù)想的用途進(jìn)行為期二個(gè)星期至三個(gè)月的短期研究
需要指出的是����,一旦臨床前研究結(jié)束��,動物試驗(yàn)并沒有結(jié)束隨之完成��,許多時(shí)間更長��、更專項(xiàng)的研究如慢性�、抗癌試驗(yàn)將在整個(gè)新藥申請過程中進(jìn)行動����。
臨床前研究用來評估:
① 藥品的藥理學(xué)現(xiàn)象和作用機(jī)理(MOA)
② 藥物毒性特征和毒性靶器官
③ 藥物吸收、分布����、代謝和排泄(ADME)
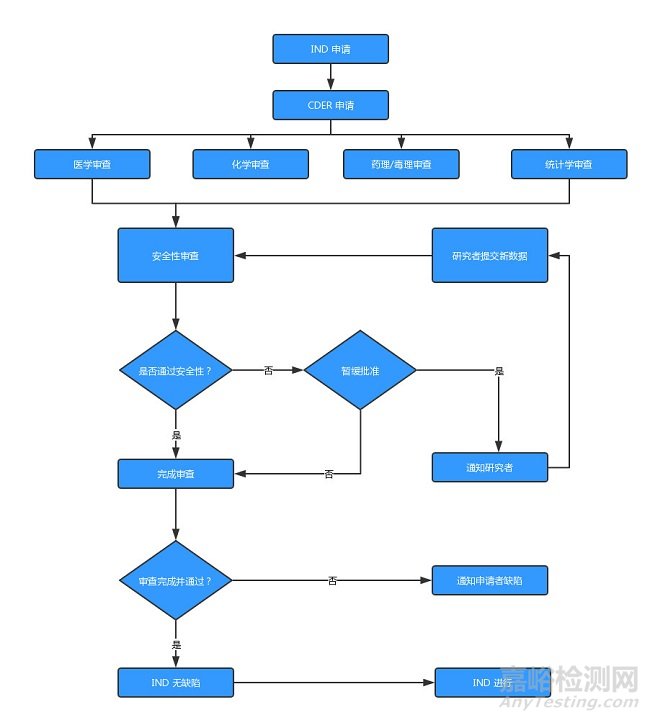
2、提出新藥臨床試驗(yàn)申請(IND)
當(dāng)申請者認(rèn)為藥品已具有足夠的數(shù)據(jù)證明該藥品是安全時(shí)�,需要向 FDA 提交新藥臨床研究申請(IND)。IND 的申請相當(dāng)于新藥申請者獲得 FDA 的許可��,可以開始在?身上進(jìn)行試驗(yàn)��。
在臨床研究申請中����,新藥申請者必須提交至少兩個(gè)領(lǐng)域的材料。首先,申請者必須向 FDA 公布所有臨床前研究的結(jié)果����,提供該新藥組成的信息,以及該新藥的生產(chǎn)與質(zhì)控程序����。
其次,申請者還必須提供臨床研究的計(jì)劃書����,并在計(jì)劃書中詳細(xì)敘述新藥申請者希望進(jìn)行的臨床研究以證明該藥用于人體的安全性、有效性����。同時(shí)�,與臨床研究相關(guān)的其它材料包括研究者資格等也必須包括在內(nèi)。
按照現(xiàn)行規(guī)定����,F(xiàn)DA 有30天的時(shí)間來決定是否允許該藥進(jìn)行人體試驗(yàn),同時(shí) FDA 還將評價(jià)臨床計(jì)劃書�。臨床研究計(jì)劃要保證臨床受試者不應(yīng)受到不必要的危險(xiǎn)以及有希望證明該新藥?于人體是安全、有效的����。
如果在提交 IND 后的30天內(nèi)����,F(xiàn)DA 沒有同新藥申請者聯(lián)系����,那么臨床試驗(yàn)就可以開始。當(dāng)然����,新藥申請者在開始臨床研究之前最好還是應(yīng)當(dāng)主動同 FDA 取得聯(lián)系。
?旦 FDA 作出決定臨床研究不應(yīng)開始�,通知會在30天內(nèi)發(fā)出,臨床試驗(yàn)會被推遲直到相關(guān)問題得到解決����。?般來說, FDA 發(fā)出「臨床試驗(yàn)暫停通知」�,主要會有以下幾個(gè)原因:
① 臨床前研究未能證明該藥?于?體會是安全的
② 臨床前研究未符合 GLP 及其它相關(guān)標(biāo)準(zhǔn)
③ 所建議的臨床研究計(jì)劃書不完整、臨床研究不安全
如果申請人在申請 IND 后的兩年內(nèi)仍沒有按計(jì)劃開始臨床研究��,或該 IND 的臨床試驗(yàn)被中止叫停超過一年�,F(xiàn)DA 便會將此 IND 列為「失活狀態(tài)」(Inactive Status)。一旦 IND 被置于「失活狀態(tài)」�,所有臨床研究者都必須被通知到,并按照21CFR 312.30的要求將臨床研究用藥品退還給申請?或立即毀。
IND分為以下兩個(gè)類別:
① 商業(yè)性 IND(Commercial IND)
商業(yè)性 IND 是指為申請新藥上市目的而申請開展的全新臨床試驗(yàn)��。IND 的申請人通常與企業(yè)進(jìn)?合作����。商業(yè)性 IND 中有?種情況被稱作「探索性 IND」(Exploratory IND)或「篩查性 IND」(Screening IND),作為遞交的第一個(gè)申報(bào)文件以支持對新藥開展最早的1期臨床研究�。
② 非商業(yè)性IND(Non-Commercial IND)
非商業(yè)性IND是由醫(yī)生自行開展的研究,該研究旨在研究藥品對特定人群的療效或?yàn)闊o藥可治的患者提供未經(jīng)批準(zhǔn)的藥物治療��。非商業(yè)性IND包括有研究性IND(InvestigatorIND/Research IND)����。
IND申報(bào)文件包主要包括9部分的內(nèi)容:
1、首頁函����、FDA 1571表
2、目錄
3����、引?和總體研究計(jì)劃
4�、研究員手冊
5、臨床研究?案
6�、化學(xué)、生產(chǎn)和質(zhì)量控制信息
7、藥理和毒理信息
8�、已有人體臨床經(jīng)驗(yàn)
9、額外信息
在IND申報(bào)文件包中�,還需提交相關(guān)原始完整研究報(bào)告,如毒理研究報(bào)告等�。
(圖:IND申請流程)
3、臨床試驗(yàn)
如果FDA批準(zhǔn)IND申請��,臨床試驗(yàn)(由人類受試者參與的研究)可以開始����。
① 1期臨床:嚴(yán)格控制藥物在少量的健康志愿者身上進(jìn)行,大約有20—80例����。這階段的試驗(yàn)主要是獲得藥物的基本的安全性數(shù)據(jù)、以及藥理信息�。受試者一般為健康志愿者。
② 2期臨床:試驗(yàn)藥物在一小部分受試者身上進(jìn)行����,大約為100到200例。這些病?是患有該藥物預(yù)設(shè)所治的疾病����。這個(gè)階段進(jìn)一步提供了該藥的安全性數(shù)據(jù)����,用于建議用途的第一個(gè)適應(yīng)癥使用該試驗(yàn)藥的有效性����。如果新藥申請者能夠從該藥的使用、或之前的臨床研究得出結(jié)果該藥用于臨床是安全的話����,?期臨床甚至某些情況下的二期臨床可以省去。
③ 3期臨床:參與受試者有數(shù)百?到數(shù)千?��,重點(diǎn)考察藥物的安全性和有效性��。試驗(yàn)藥物在較多的受試者之間進(jìn)行��,這些受試者患有該藥物預(yù)設(shè)所治��、診斷�、預(yù)防等的疾病。在開始本階段研究之前����,新藥申請者必須向 FDA 提交從一�、二期臨床試驗(yàn)中的數(shù)據(jù)以表明該藥是有理由安全����、有效的以及具有有利的效益/風(fēng)險(xiǎn)比����。
關(guān)于 End of Phase IIA meeting(EOP2A)會議
① 在II期臨床試驗(yàn)完成以后,F(xiàn)DA強(qiáng)烈建議申請人在開始III期關(guān)鍵性臨床試驗(yàn)之前�,提出 EOP2 會議。EOP2A 會議發(fā)生在臨床試驗(yàn)獲得在擬定適應(yīng)癥中的劑量反應(yīng)關(guān)系后����,包括劑量范圍對安全性、生物標(biāo)志物和概念性驗(yàn)證的影響����;通常發(fā)生在I期臨床試驗(yàn)完成和第?批患者暴露-反應(yīng)試驗(yàn)之后和IIb期(如患者劑量范圍試驗(yàn))和III期臨床有效性安全性試驗(yàn)之前。
② EOP2A 有助于幫助申請人找到最佳劑量��,節(jié)約成本�,將后期臨床試驗(yàn)的成功率最?化。為了能夠充分利用 EOP2 會議與 FDA 進(jìn)?溝通��,在會議開始一個(gè)月之前��,申請人應(yīng)向 FDA 提交一個(gè)會議文件包:
1��、會議請求與會議信息
2、日期�、時(shí)間和參會者
3、總結(jié)所有更新的數(shù)據(jù)(臨床, CMC, 藥理/毒理等)
4�、提出 Phase III 開發(fā)計(jì)劃
5、提出可能使?的藥物標(biāo)
6�、問答
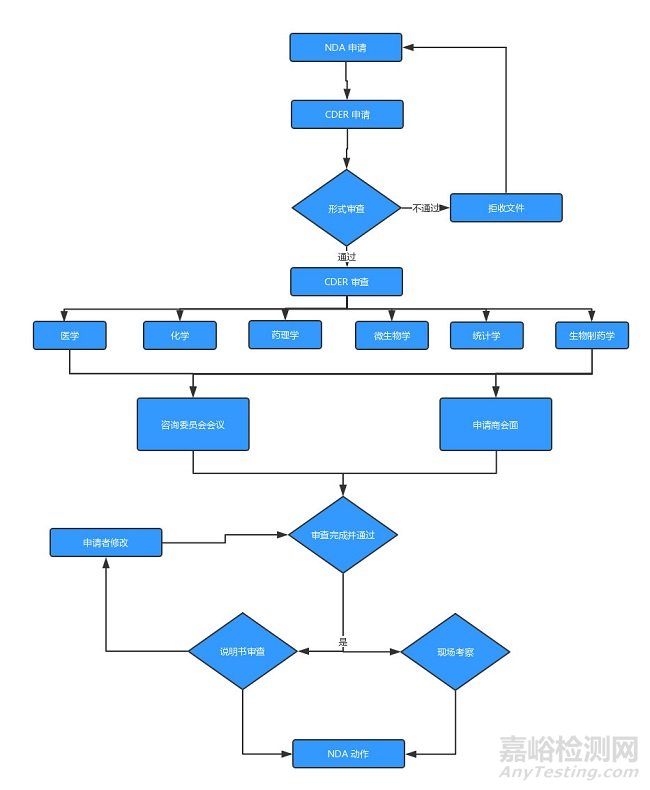
4、新藥申請(NDA)
臨床試驗(yàn)結(jié)束之后��,藥物申請者可提交一份 NDA��,申請批準(zhǔn)這款藥物在美國銷售�。FDA 根據(jù)藥品的治療特性,在審查程序上分為「標(biāo)準(zhǔn)審查(Standard Review�,SR)」和「優(yōu)先審查(Priority Review,PR)」兩類����。
對「能夠在治療、診斷或疾病預(yù)防上?已上市藥品有顯著改進(jìn)的新藥」��,F(xiàn)DA 對非常重要的 NDAs 在6個(gè)?內(nèi)進(jìn)性行審評�,新藥的標(biāo)準(zhǔn)審評時(shí)間是10個(gè)月。FDA根據(jù)《政策和程序?冊》(Manual of Policiesand Procedures��,MAPPs)相關(guān)規(guī)定可采取「優(yōu)先審查」(Priority Review)����,評審時(shí)間由標(biāo)準(zhǔn)審查的10個(gè)月縮短為6個(gè)月。NDA評審是最嚴(yán)格�、耗時(shí)的過程,而且只有很小比例的試驗(yàn)藥最終能允許進(jìn)入了市場����。
此外,F(xiàn)DA還通過快速通道(FastTrack)來鼓勵(lì)藥物創(chuàng)新和加快審查?于治療嚴(yán)重或威脅?命疾病或尚未滿?臨床治療需求的新藥��,例如艾滋病����、阿爾茲海默病、心衰����、腫瘤、癲癇��、抑郁癥和糖尿病等��。一旦符合快速通道(Fast Track)的藥品��,F(xiàn)DA必須在60天內(nèi)做出決策����。
(圖:INA申請流程)
5.新藥上市后的監(jiān)測
藥物被批準(zhǔn)之后�,藥品的標(biāo)簽可能進(jìn)行變更�,內(nèi)容包括藥物副作用的新信息。藥物申請者需要提交安全性變更����,醫(yī)?或患者也可以向 FDA報(bào)告有關(guān)藥物的嚴(yán)重不良事件。引起更嚴(yán)重��、超出預(yù)期副作用的藥物在必要的情況下要從市場撤市�。
6.NDA 申請費(fèi)用 PDUFA
PDUFA 即《處方藥申報(bào)者付費(fèi)法案》,F(xiàn)DA 依據(jù)該法案向制藥商/申報(bào)者收取一定的審查費(fèi)用�。PDUFA 費(fèi)用主要包括三部分:
1、申請費(fèi)
2��、生產(chǎn)設(shè)施費(fèi)
3�、產(chǎn)品費(fèi)
法案同時(shí)也規(guī)定,PDUFA 費(fèi)用只針對新藥申請階段(NDA)收取有關(guān)費(fèi)用��,而對于臨床前研究(PreIND)申報(bào)資料的審評��,是不收取有關(guān)費(fèi)用的��。
費(fèi)用的免除、削減和退還PDUFA 條款中也指出�,當(dāng)免除費(fèi)用的決定有助于公眾健康和安全、費(fèi)用收取會給藥品創(chuàng)新帶來障礙�、或者所收費(fèi)用超過審查成本時(shí),F(xiàn)DA 可做出免除�、削減或退還收費(fèi)的決定。
References:
[1]《讀懂 FDA 藥品注冊》
[2]《如何獲得 FDA 批準(zhǔn)》
[3]《FDA 官方網(wǎng)站》