藥品上市后變更管理是藥品全生命周期管理的重要組成部分����,其監(jiān)管也是各國(guó)或地區(qū)藥品監(jiān)管的重點(diǎn)和難點(diǎn)�。原料藥是藥品的活性成分,在預(yù)防�、診斷、處置�、緩解或治療疾病中發(fā)揮藥理作用或其它直接效用,或者影響人體的結(jié)構(gòu)和機(jī)能���。所以����,原料藥的質(zhì)量直接決定了藥品的質(zhì)量,原料藥的上市后變更也直接對(duì)藥品的安全性和有效性產(chǎn)生影響�。
原料藥上市后變更應(yīng)該以其注冊(cè)上市時(shí)或登記時(shí)開展的研究驗(yàn)證工作,以及既往實(shí)際生產(chǎn)過程中積累的數(shù)據(jù)和上市后研究為基礎(chǔ)���,原料藥生產(chǎn)企業(yè)對(duì)原料藥本身的了解程度決定了其在預(yù)計(jì)發(fā)生變更時(shí)對(duì)風(fēng)險(xiǎn)的預(yù)知程度����,而對(duì)風(fēng)險(xiǎn)的預(yù)知程度就決定了其進(jìn)行變更研究的廣度和深度���,從而最終決定了所實(shí)施的變更對(duì)產(chǎn)品所產(chǎn)生的實(shí)際影響���。因此���,原料藥上市后的變更評(píng)估就顯得尤為重要���,它是企業(yè)充分認(rèn)識(shí)和總結(jié)品種特點(diǎn)、預(yù)知變更可能存在風(fēng)險(xiǎn)及針對(duì)風(fēng)險(xiǎn)制定研究和驗(yàn)證內(nèi)容的系統(tǒng)性工作���。
一����、原料藥變更的管理要求
各國(guó)/地區(qū)對(duì)原料藥的法律界定不盡相同,但藥品監(jiān)管體系較為成熟的國(guó)家和地區(qū)����,都將原料藥上市后變更作為藥品監(jiān)管的重點(diǎn)。美國(guó)食品藥品管理局(FDA)雖然對(duì)原料藥主要采取登記管理�,即藥物主文件(DMF)制度,但FDA的法規(guī)對(duì)藥品定義了三個(gè)級(jí)別的變更上報(bào)類型����,對(duì)登記原料藥同樣適用,對(duì)于在制劑申請(qǐng)中提供原料藥信息的�,通過制劑上報(bào)變更,即需事先批準(zhǔn)的主要變更補(bǔ)充申請(qǐng)����、30天生效或立即生效的中等變更補(bǔ)充申請(qǐng)、在年度報(bào)告(AR)中上報(bào)的微小變更�。
原料藥以前在我國(guó)獲批后有相應(yīng)的藥品批準(zhǔn)文號(hào),相關(guān)上市后變更自然也是按藥品上市后變更管理���。自2017年11月起���,我國(guó)對(duì)原料藥采取登記形式,不再頒發(fā)藥品批準(zhǔn)文號(hào)����,但原料藥的審批依舊是行政許可事項(xiàng)���,雖然原料藥采取關(guān)聯(lián)審評(píng)審批,但實(shí)際管理依舊是按照藥品管理����。《藥品上市后變更管理辦法(試行)》中也明確原料藥發(fā)生變更時(shí)要按照相關(guān)管理規(guī)定及技術(shù)指導(dǎo)原則����,進(jìn)行充分研究、評(píng)估和必要的驗(yàn)證�,確定變更管理類別,并按法規(guī)要求對(duì)于生產(chǎn)過程中的重大變更需要報(bào)補(bǔ)充申請(qǐng)經(jīng)國(guó)家局批準(zhǔn)后實(shí)施�,中等變更需要在所在省局備案后實(shí)施,微小變更實(shí)施后需要在年度報(bào)告中報(bào)告�。
二���、原料藥生產(chǎn)過程變更研究評(píng)估
相對(duì)于同一劑型的制劑生產(chǎn)工藝比較類似的情況����,原料藥的生產(chǎn)涉及的化學(xué)反應(yīng)有成千上萬種���,所以原料藥變更的研究及評(píng)估更加復(fù)雜和困難�。因?yàn)樵纤幧a(chǎn)工藝更具有特異性,所以對(duì)于原料藥生產(chǎn)過程的變更�,結(jié)合品種特點(diǎn)的要求就更為突出。
在這種情況下���,各國(guó)的原料藥變更指導(dǎo)原則都不可能將原料藥可能涉及的變更情形一一列舉����。因此更需要生產(chǎn)企業(yè)全面掌握所生產(chǎn)品種的特點(diǎn)�、工藝控制的要求,根據(jù)指導(dǎo)原則精神和風(fēng)險(xiǎn)控制的目的���,以及提出的變更總體考慮����、變更研究驗(yàn)證的思路�,對(duì)計(jì)劃發(fā)生的變更進(jìn)行深層次的原因和可能存在的風(fēng)險(xiǎn)進(jìn)行分析,進(jìn)而開展相應(yīng)的研究和驗(yàn)證工作����,以此達(dá)到變更后能持續(xù)生產(chǎn)出質(zhì)量不降低的原料藥的目標(biāo)。對(duì)于原料藥生產(chǎn)工藝變更的評(píng)估應(yīng)該系統(tǒng)全面,充分考慮變更對(duì)后續(xù)反應(yīng)及反應(yīng)后處理的影響和潛在風(fēng)險(xiǎn)����,不能僅僅局限于變更步驟或操作本身,這樣才能達(dá)到有效控制風(fēng)險(xiǎn)的目的�。
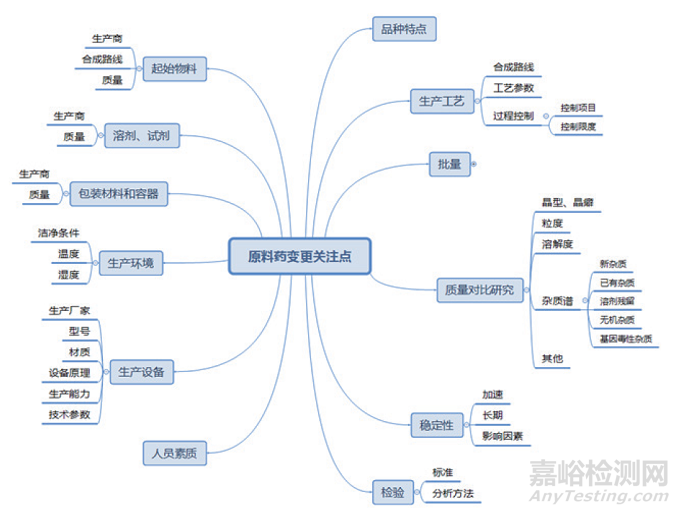
本文所討論的原料藥變更,僅涉及合成原料藥以及半合成原料藥的合成步驟及之后的生產(chǎn)工藝�。這也是美國(guó)《原料藥批準(zhǔn)后變更行業(yè)指南》和我國(guó)《已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》中變更原料藥生產(chǎn)工藝部分的適用范圍?��;瘜W(xué)合成原料藥發(fā)生變更時(shí)���,可能影響原料藥質(zhì)量的因素有很多,如圖所示����,所以企業(yè)需要在變更時(shí)對(duì)諸多因素進(jìn)行綜合評(píng)估。
原料藥變更影響因素思維導(dǎo)圖
品種特點(diǎn)方面���。無論是制劑還是原料藥����,充分了解品種特點(diǎn)是有效控制變更風(fēng)險(xiǎn)的基礎(chǔ)����。對(duì)于原料藥的品種特點(diǎn),應(yīng)從反應(yīng)條件�、化合物的結(jié)構(gòu)以及化合物的穩(wěn)定性等方面進(jìn)行考慮。這里考慮的化合物不只是最終的原料藥���,還包括起始物料���、合成過程中的中間體或中間產(chǎn)物。如果存在化合物或反應(yīng)過程中使用的試劑對(duì)光���、水���、氧等敏感,在相應(yīng)的步驟或反應(yīng)后處理中應(yīng)考慮反應(yīng)過程中及相應(yīng)操作時(shí)的控制措施���。如果變更后的化合物穩(wěn)定性差的���,應(yīng)根據(jù)其降解或變化的可能,在生產(chǎn)工藝和中間體保存中采取必要的措施�,以達(dá)到能持續(xù)穩(wěn)定地獲得目標(biāo)化合物和控制有關(guān)物質(zhì)的目的。
人員素質(zhì)方面����。經(jīng)驗(yàn)豐富的人員在同一設(shè)施中實(shí)施變更的風(fēng)險(xiǎn)����,會(huì)比沒有經(jīng)驗(yàn)的人員實(shí)施變更的風(fēng)險(xiǎn)小����。即便是設(shè)備也發(fā)生了變更,既往有過使用相同或類似設(shè)備經(jīng)驗(yàn)的人員也比未使用過的人員實(shí)施變更的風(fēng)險(xiǎn)小���。對(duì)于原料藥�,人員如果曾經(jīng)有過操作類似化學(xué)合成反應(yīng)經(jīng)驗(yàn)的也比無相關(guān)經(jīng)驗(yàn)人員的風(fēng)險(xiǎn)小����,特別是對(duì)于原料藥合成中反應(yīng)條件苛刻、反應(yīng)過程劇烈或反應(yīng)中有燃燒或爆炸可能性的反應(yīng)���,相關(guān)經(jīng)驗(yàn)就更為重要����。
設(shè)施設(shè)備方面�。在合成路線不變的前提下,設(shè)施設(shè)備的變更評(píng)估應(yīng)重點(diǎn)關(guān)注設(shè)備變更發(fā)生的工序�。如果是因合成路線等變更引起的設(shè)施設(shè)備的變更����,應(yīng)按照關(guān)聯(lián)變更的要求進(jìn)行相應(yīng)的評(píng)估和研究驗(yàn)證����。如果設(shè)施設(shè)備變更是最后一步反應(yīng)之后不同設(shè)計(jì)和工作原理的設(shè)備變更���,則更容易引起原料藥物理性質(zhì)的變化����。設(shè)備材質(zhì)變更時(shí)要考慮新材質(zhì)與反應(yīng)的相容性����。生產(chǎn)規(guī)模的變更往往關(guān)聯(lián)設(shè)備變更,如果僅生產(chǎn)批量變更����,那么設(shè)備變更應(yīng)為相同材料、設(shè)計(jì)及操作原理����,僅是容量上的不同,且工藝參數(shù)的調(diào)整應(yīng)限于適應(yīng)設(shè)備變化需求���,并且關(guān)鍵工藝參數(shù)應(yīng)在申報(bào)范圍之內(nèi)����。但在原料藥批量變化較大時(shí),研究工作總體上應(yīng)按照技術(shù)要求較高的變更類別進(jìn)行���。
雜質(zhì)方面���。在原料藥生產(chǎn)過程的變更中,雜質(zhì)的研究不僅僅是對(duì)質(zhì)量標(biāo)準(zhǔn)中規(guī)定的雜質(zhì)進(jìn)行檢驗(yàn)后比較����,因?yàn)樯a(chǎn)工藝的變更往往會(huì)引起原料藥雜質(zhì)情況的變化。在評(píng)估時(shí)要綜合考慮已有雜質(zhì)���、新雜質(zhì)����、殘留溶劑���、無機(jī)雜質(zhì)�、基因毒雜質(zhì)的變化情況�。如果變更后原料藥的已有雜質(zhì)及雜質(zhì)總量與歷史數(shù)據(jù)相當(dāng)�,可以視為雜質(zhì)譜一致����。變更前后雜質(zhì)譜一致的,變更風(fēng)險(xiǎn)較小���,雜質(zhì)譜不一致,變更風(fēng)險(xiǎn)較高�。評(píng)估變更對(duì)雜質(zhì)譜的影響應(yīng)根據(jù)實(shí)際情況確定在哪個(gè)工藝階段評(píng)估雜質(zhì),非分離的物料一般不適合用于證明雜質(zhì)譜的一致性����,應(yīng)在變更所在步驟之后的某已分離物料的雜質(zhì)譜與變更前對(duì)比研究。
物理性質(zhì)方面�。原料藥生產(chǎn)工藝的變更,特別是原料藥最后一個(gè)溶液步驟及其后步驟的變更最有可能影響原料藥的物理性質(zhì)���。對(duì)于最終以溶液劑型給藥或制劑生產(chǎn)過程中原料藥需要完全溶解的劑型及快速溶解的口服劑型等�,理化性質(zhì)的變更對(duì)制劑的影響比較小�。但對(duì)于難溶原料藥的口服制劑、原料藥為固體的非口服劑型����、受原料藥物理性質(zhì)影響顯著的劑型���、局部用藥的粉末劑型、部分改良釋放速度的藥品等����,物理性質(zhì)就需要特別關(guān)注。變更發(fā)生在原料藥最終沉淀�、原料藥部分溶解的打漿等步驟時(shí),對(duì)原料藥物理性質(zhì)的變化產(chǎn)生影響的可能性更高���。設(shè)施設(shè)備和生產(chǎn)批量的變更���,也可能會(huì)對(duì)原料藥的物理性質(zhì)產(chǎn)生影響。
藥品質(zhì)量關(guān)系到人民群眾的身體健康和生命安全���,原料藥的質(zhì)量直接決定了藥品的質(zhì)量����。原料藥登記人在實(shí)施變更過程中���,應(yīng)結(jié)合品種特點(diǎn)�,依據(jù)申報(bào)注冊(cè)的工藝研究和既往生產(chǎn)的歷史數(shù)據(jù)����,充分評(píng)估變更可能存在的風(fēng)險(xiǎn)���,并針對(duì)這些潛在風(fēng)險(xiǎn)開展相關(guān)研究驗(yàn)證工作,從而保證變更后的原料藥質(zhì)量不降低�、不對(duì)相關(guān)制劑產(chǎn)生不良影響。制劑的持有人也應(yīng)切實(shí)履行主體責(zé)任���,按法規(guī)及自身生產(chǎn)要求對(duì)原料藥供應(yīng)商進(jìn)行規(guī)范管理,及時(shí)了解原料藥的變更情況���,從而保證藥品的安全����、有效和質(zhì)量可控����。