為了鼓勵(lì)以臨床價(jià)值為導(dǎo)向的藥物創(chuàng)新,現(xiàn)行《藥品注冊(cè)管理辦法》明確提出了4種加快上市注冊(cè)的程序��。對(duì)于早期臨床試驗(yàn)數(shù)據(jù)可證實(shí)藥物療效并能預(yù)測(cè)其臨床價(jià)值的創(chuàng)新藥�����,經(jīng)評(píng)價(jià)后可以通過(guò)附條件批準(zhǔn)工作程序���,在完成確證性臨床研究之前上市��,讓有嚴(yán)重危及生命且尚無(wú)治療手段的患者獲得更多延長(zhǎng)其生命或改善其生活質(zhì)量的治療機(jī)會(huì)���。附條件批準(zhǔn)工作程序可以縮短藥品上市前的臨床研發(fā)時(shí)間�����,因此其配套政策和審評(píng)標(biāo)準(zhǔn)備受關(guān)注。
本文以腫瘤創(chuàng)新藥為切入點(diǎn)���,對(duì)我國(guó)附條件批準(zhǔn)的政策要求和審評(píng)過(guò)程中已經(jīng)形成的標(biāo)準(zhǔn)進(jìn)行系統(tǒng)的梳理�����,并嘗試對(duì)附條件批準(zhǔn)實(shí)施過(guò)程中遇到的問(wèn)題進(jìn)行分析并提出建議��,供業(yè)界參考���。
現(xiàn)行《藥品注冊(cè)管理辦法》(下文稱“辦法”)于2020年7月1日起正式實(shí)施[1]。與2007版辦法相比��,現(xiàn)行辦法最大的變化之一是明確提出了4種藥品加快上市注冊(cè)程序���,即突破性治療藥物��、附條件批準(zhǔn)�����、優(yōu)先審評(píng)審批及特別審批程序��,鼓勵(lì)以臨床價(jià)值為導(dǎo)向的藥物創(chuàng)新���。
對(duì)于被列入加快上市注冊(cè)程序的藥品��,藥品監(jiān)督管理部門及專業(yè)技術(shù)機(jī)構(gòu)將給予政策和技術(shù)支持��,提供更多的技術(shù)指導(dǎo)��,優(yōu)先配置溝通交流和審評(píng)資源并盡可能縮短審評(píng)時(shí)限���。4種加快上市注冊(cè)程序中,附條件批準(zhǔn)直接以縮短藥物臨床試驗(yàn)的研發(fā)時(shí)間為目的�����,其配套政策和審批標(biāo)準(zhǔn)最受業(yè)界關(guān)注�����。在附條件批準(zhǔn)的適用情形中,“治療嚴(yán)重危及生命且尚無(wú)有效治療手段疾病的藥品”相比其他2種情形的潛在對(duì)象相對(duì)廣泛���,其臨床研究的設(shè)計(jì)要求和審評(píng)標(biāo)準(zhǔn)更易成為持續(xù)性焦點(diǎn)�����,監(jiān)管挑戰(zhàn)也更大�����。
現(xiàn)行辦法實(shí)施僅2年余,但在此之前已有多個(gè)國(guó)產(chǎn)抗腫瘤(包括血液系統(tǒng)惡性疾?��。﹦?chuàng)新藥基于附條件批準(zhǔn)的理念獲批上市; 在附條件批準(zhǔn)正式成為中國(guó)加快上市注冊(cè)程序之一后��,腫瘤也是附條件批準(zhǔn)藥品中占比最高的適應(yīng)證類型���。因此,腫瘤適應(yīng)證是附條件批準(zhǔn)的先行者和積極踐行者���,也是監(jiān)管思考相對(duì)成熟的適應(yīng)證�����。
本文以腫瘤創(chuàng)新藥為切入點(diǎn)��,對(duì)我國(guó)附條件批準(zhǔn)的政策要求和審評(píng)過(guò)程中已經(jīng)形成的標(biāo)準(zhǔn)進(jìn)行系統(tǒng)的梳理�����,供業(yè)界參考���。本文亦嘗試對(duì)附條件批準(zhǔn)實(shí)施過(guò)程中監(jiān)管部門面臨的關(guān)鍵挑戰(zhàn)進(jìn)行分析并提出建議��。
1���、我國(guó)附條件批準(zhǔn)的基本涵義
現(xiàn)行辦法明確了可申請(qǐng)附條件批準(zhǔn)的3種情形,并且對(duì)附條件批準(zhǔn)的申請(qǐng)���、流程���、上市后應(yīng)履行的“條件”提出了要求。在具體實(shí)施過(guò)程中��,可參考的技術(shù)性文件還包括國(guó)家藥品監(jiān)督管理局2020年7月7日發(fā)布的《藥品附條件批準(zhǔn)上市申請(qǐng)審評(píng)審批工作程序(試行)》[2](下文稱“工作程序”)和國(guó)家藥品監(jiān)督管理局藥品審評(píng)中心2020年11月19日發(fā)布的《藥品附條件批準(zhǔn)上市技術(shù)指導(dǎo)原則(試行)》[3]��。
實(shí)施附條件批準(zhǔn)上市的目的在于“縮短藥物臨床試驗(yàn)的研發(fā)時(shí)間,使其盡早應(yīng)用于無(wú)法繼續(xù)等待的危重疾病或公共衛(wèi)生方面急需的患者”[3]���。在可獲得疾病病理生理特征���、藥物機(jī)制、臨床前安全性和藥效學(xué)研究結(jié)果支持的前提下��,若藥物已有臨床研究數(shù)據(jù)所反映的治療獲益已經(jīng)足夠大���,以至于目標(biāo)適應(yīng)證人群繼續(xù)等待常規(guī)批準(zhǔn)所需要的臨床研究證據(jù)所面臨的生命或健康損失���,極有可能遠(yuǎn)超因臨床獲益尚未得到確證帶來(lái)的風(fēng)險(xiǎn),從而得出“藥物臨床試驗(yàn)已有數(shù)據(jù)可證實(shí)藥物療效并能預(yù)測(cè)其臨床價(jià)值”的結(jié)論[1]�����,則可在藥物完成確證性臨床試驗(yàn)前基于替代終點(diǎn)�����、中間臨床終點(diǎn)或早期臨床試驗(yàn)數(shù)據(jù)批準(zhǔn)藥物上市�����。
附條件批準(zhǔn)并不意味著降低新藥上市的技術(shù)要求���。遞交附條件批準(zhǔn)上市許可申請(qǐng)的腫瘤創(chuàng)新藥物�����,藥學(xué)��、藥理毒理學(xué)要求與常規(guī)批準(zhǔn)上市藥品相同��,申請(qǐng)人也不應(yīng)該以“開展上市后研究”作為彌補(bǔ)臨床試驗(yàn)設(shè)計(jì)或數(shù)據(jù)缺陷的條件提出附條件批準(zhǔn)申請(qǐng)��。藥物在獲得附條件批準(zhǔn)上市后��,上市許可持有人(簡(jiǎn)稱“持有人”)應(yīng)繼續(xù)按照常規(guī)批準(zhǔn)的要求開展確證性臨床試驗(yàn)���,以全面支持藥物的獲益風(fēng)險(xiǎn)評(píng)價(jià)。
全球范圍內(nèi)已有多個(gè)藥品監(jiān)管機(jī)構(gòu)為鼓勵(lì)危重疾病領(lǐng)域的藥物創(chuàng)新���,向工業(yè)界提供加快注冊(cè)程序���。表1將我國(guó)的“附條件批準(zhǔn)”與美國(guó)、歐盟和日本藥品監(jiān)管機(jī)構(gòu)實(shí)施的類似程序進(jìn)行了比較。雖然各國(guó)監(jiān)管機(jī)構(gòu)在技術(shù)細(xì)節(jié)上存在差異�����,但整體原則是相似的�����,即對(duì)于嚴(yán)重威脅生命或患者生活質(zhì)量的疾病���,如果新藥現(xiàn)有數(shù)據(jù)呈現(xiàn)良好的獲益風(fēng)險(xiǎn)比且可預(yù)測(cè)其臨床價(jià)值��,可以在完成一般新藥注冊(cè)上市所需要的確證性研究前提前獲批上市���。

2、境內(nèi)獲附條件批準(zhǔn)的抗腫瘤藥品
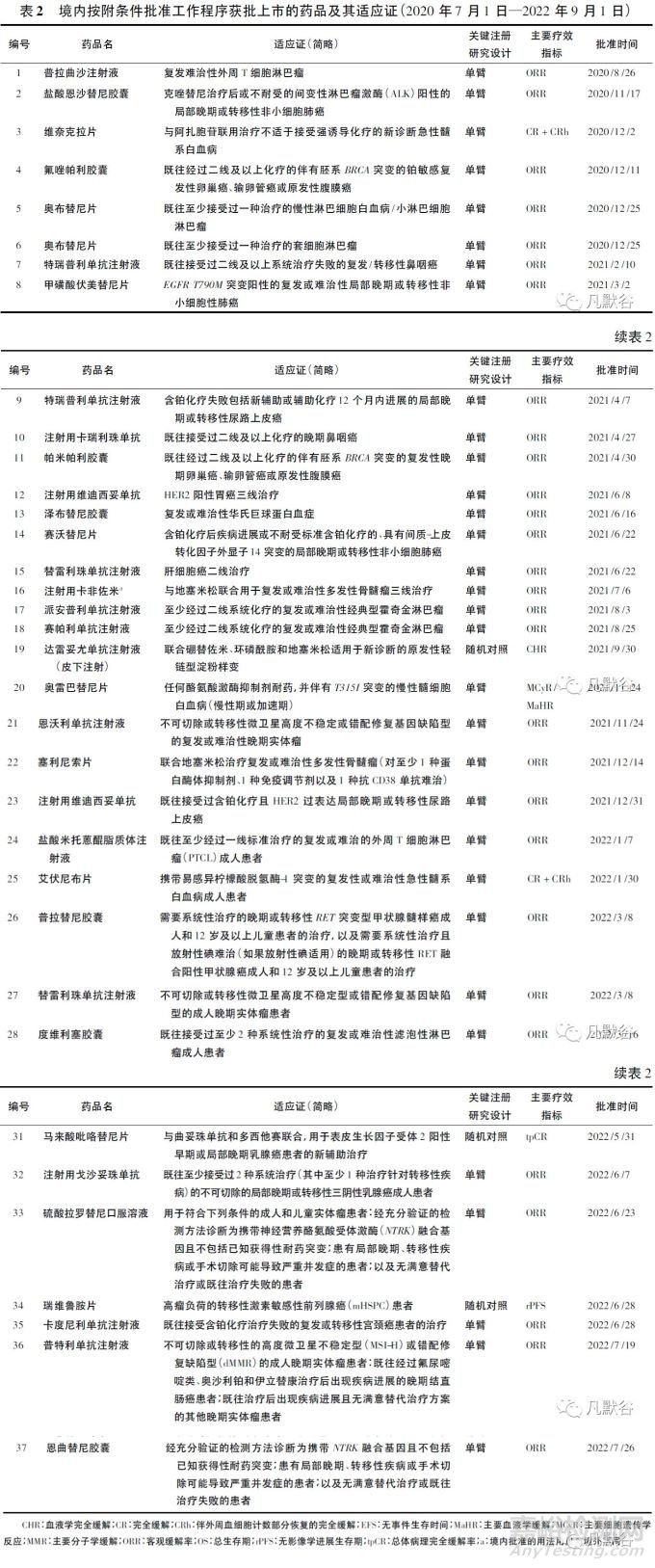
自現(xiàn)行辦法實(shí)施以來(lái)�����,截至2022年9月1日�����,已有32個(gè)化學(xué)藥品或單抗類生物制品的37個(gè)腫瘤適應(yīng)證在境內(nèi)獲附條件批準(zhǔn)上市(見表2)�����。在附條件批準(zhǔn)工作程序正式落地之前��,西達(dá)本胺��、吡咯替尼���、信迪利單抗��、特瑞普利單抗、卡瑞利珠單抗和鹽酸安羅替尼等藥品已通過(guò)與現(xiàn)行“附條件批準(zhǔn)”相似的研發(fā)策略獲得首個(gè)適應(yīng)證的批準(zhǔn)��。
在現(xiàn)行辦法實(shí)施初期的過(guò)渡階段��,部分境外已上市的藥品���,因其境外數(shù)據(jù)顯示出突出的治療獲益���,中國(guó)患者臨床需求迫切且存在種族敏感性的可能性極小,在中國(guó)受試者數(shù)據(jù)非常有限時(shí)或者完全基于境外數(shù)據(jù)獲得“附條件批準(zhǔn)”�����。這些藥品實(shí)質(zhì)上是沿用了現(xiàn)行辦法實(shí)施前的“有條件批準(zhǔn)”,即上市后須在中國(guó)受試者中補(bǔ)充開展臨床研究�����,其本質(zhì)與現(xiàn)行辦法中定義的“附條件批準(zhǔn)”存在區(qū)別���。
對(duì)附條件批準(zhǔn)的抗腫瘤藥品關(guān)鍵性注冊(cè)研究設(shè)計(jì)思路進(jìn)行總結(jié)�����,可以分為以下3種模式:
① 單臂研究注冊(cè):絕大多數(shù)附條件批準(zhǔn)的藥品在無(wú)治療手段的末線腫瘤患者中開展單臂設(shè)計(jì)的關(guān)鍵性注冊(cè)研究��,最常用的替代終點(diǎn)是客觀緩解率(ORR)相比歷史數(shù)據(jù)的突出改善���。在以緩解率作為主要療效指標(biāo)的單臂設(shè)計(jì)研究數(shù)據(jù)中���,緩解持續(xù)時(shí)間是評(píng)價(jià)緩解質(zhì)量的關(guān)鍵指標(biāo),也是通過(guò)緩解率合理預(yù)測(cè)臨床獲益的重要支持性數(shù)據(jù)��。
② 小樣本量Ⅱ期隨機(jī)對(duì)照研究注冊(cè):相比單臂研究,以替代終點(diǎn)作為主要研究終點(diǎn)開展小樣本量的隨機(jī)對(duì)照研究可以更直接地反映藥品的治療獲益�����。吡咯替尼即是基于一項(xiàng)包括128例復(fù)發(fā)或轉(zhuǎn)移性乳腺癌患者的Ⅱ期臨床試驗(yàn)結(jié)果[8]�����,獲得了“聯(lián)合卡培他濱治療表皮生長(zhǎng)因子受體2(HER2)陽(yáng)性��、蒽環(huán)類或紫杉類化療治療失敗的復(fù)發(fā)或轉(zhuǎn)移性乳腺癌患者”適應(yīng)證的附條件批準(zhǔn)�����。該研究以拉帕替尼聯(lián)合卡培他濱方案作為對(duì)照���,在末例受試者至少接受12個(gè)周期治療時(shí)���,試驗(yàn)組和對(duì)照組的主要療效指標(biāo)即研究者評(píng)估的ORR分別為78.5%和57.1%; 試驗(yàn)組相比對(duì)照組的無(wú)進(jìn)展生存時(shí)間[中位值分別為18.1和7.0個(gè)月,風(fēng)險(xiǎn)比(HR)=0.363]和總生存期[尚未成熟,中位值分別為未達(dá)到和16.2個(gè)月,HR=0.362(95%CI: 0.160~0.815)]優(yōu)勢(shì)�����,為附條件批準(zhǔn)提供了非常有價(jià)值的支持性數(shù)據(jù)��。
③ 確證性研究早期數(shù)據(jù)注冊(cè):關(guān)鍵性注冊(cè)研究采用適應(yīng)性設(shè)計(jì)�����,以替代終點(diǎn)或期中分析結(jié)果提出附條件批準(zhǔn)申請(qǐng)��,再以臨床終點(diǎn)主要分析結(jié)果支持常規(guī)批準(zhǔn),是一種更為穩(wěn)妥和高效的注冊(cè)策略��。全球Ⅲ期臨床研究AMY3001中���,達(dá)雷妥尤單抗皮下注射聯(lián)合標(biāo)準(zhǔn)治療CyBorD(環(huán)磷酰胺�����、硼替佐米���、地塞米松)方案與CyBorD方案相比��,主要療效指標(biāo)血液學(xué)完全緩解率得到了顯著改善(53.3%vs18.1%); 中位隨訪11.4個(gè)月時(shí)���,主要器官衰竭無(wú)進(jìn)展生存期(MOD-PFS)期中分析結(jié)果表明試驗(yàn)組相比對(duì)照組HR=0.580(95%CI: 0.363~0.926��,兩組均未達(dá)到中位值),主要器官衰竭無(wú)事件生存期(中位值分別為未達(dá)到vs8.8個(gè)月,HR=0.39,95%CI: 0.27~0.56)也得到了顯著的延長(zhǎng)[9]�����。
基于上述研究結(jié)果���,達(dá)雷妥尤單抗(皮下制劑)獲得了“新診斷的原發(fā)性輕鏈型淀粉樣變”適應(yīng)證的附條件批準(zhǔn)��。由于MOD-PFS分析為預(yù)設(shè)的中期分析(計(jì)劃事件的43.5%)�����,名義P值(0.0211)未越過(guò)預(yù)先規(guī)定的終止界值�����,因此研究還將繼續(xù)進(jìn)行���,以MOD-PFS的最終分析結(jié)果以及更成熟的總生存期數(shù)據(jù)支持常規(guī)批準(zhǔn)[9]�����。
3���、附條件批準(zhǔn)溝通及審評(píng)過(guò)程中的關(guān)注重點(diǎn)
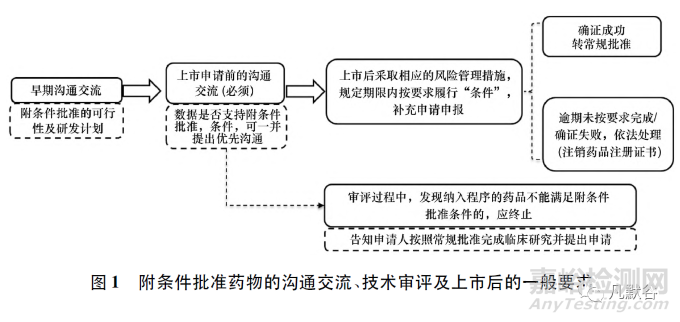
相比常規(guī)批準(zhǔn)���,附條件批準(zhǔn)在前期溝通、技術(shù)審評(píng)和上市后要求方面必然有所不同(見圖1)[2-3]���。附條件批準(zhǔn)的適用性和技術(shù)要求可能受目標(biāo)適應(yīng)證的特征和藥物的創(chuàng)新程度影響�����,并且隨臨床實(shí)踐的變化而發(fā)生實(shí)質(zhì)性的改變���,凸顯了溝通交流的重要性。
在抗腫瘤創(chuàng)新藥附條件批準(zhǔn)的溝通交流中�����,應(yīng)該重點(diǎn)解決以下問(wèn)題:
① 目標(biāo)適應(yīng)證所對(duì)應(yīng)的附條件批準(zhǔn)適用情形���。多數(shù)腫瘤有相對(duì)標(biāo)準(zhǔn)的治療路徑,還有部分腫瘤因?yàn)榘邢蛩幬锏某霈F(xiàn)呈慢病化趨勢(shì)�����,申請(qǐng)人應(yīng)該分析目標(biāo)適應(yīng)證當(dāng)前最亟待解決的臨床問(wèn)題,結(jié)合藥物的作用機(jī)制和已有臨床數(shù)據(jù)所反映的潛在優(yōu)勢(shì)定位目標(biāo)人群���,是無(wú)有效治療手段的復(fù)發(fā)難治患者還是已有標(biāo)準(zhǔn)方案的新診斷患者��,或是采用適當(dāng)?shù)纳飿?biāo)志物篩選耐藥或預(yù)后更差的亞組�����,不同的人群定位將對(duì)應(yīng)不同的附條件批準(zhǔn)技術(shù)要求���,對(duì)研發(fā)計(jì)劃的制定產(chǎn)生重要影響。
② 支持附條件批準(zhǔn)關(guān)鍵性注冊(cè)研究的設(shè)計(jì)思路��。單臂研究通常適用于經(jīng)現(xiàn)有治療手段充分治療后反應(yīng)不佳或多次復(fù)發(fā)�����,且無(wú)有效治療手段的末線腫瘤患者���。如果目標(biāo)人群有標(biāo)準(zhǔn)治療或推薦治療方案��,采用隨機(jī)對(duì)照研究驗(yàn)證試驗(yàn)藥/方案相比現(xiàn)有治療手段的優(yōu)勢(shì)是更適宜的設(shè)計(jì)思路�����。
③ 替代終點(diǎn)是否“很可能預(yù)測(cè)臨床獲益”��,或擬采用的中間臨床終點(diǎn)能否預(yù)測(cè)長(zhǎng)期臨床獲益���。申請(qǐng)人可以通過(guò)相同腫瘤類型的既往數(shù)據(jù)進(jìn)行論證���,但需要注意藥物的作用機(jī)制也可能對(duì)替代終點(diǎn)/中間終點(diǎn)與臨床終點(diǎn)之間的相關(guān)性或相關(guān)程度產(chǎn)生影響。例如: 通常認(rèn)為腫瘤縮小的影像學(xué)證據(jù)(客觀緩解)和緩解持續(xù)時(shí)間能合理預(yù)測(cè)總生存期的改善���,但免疫檢查點(diǎn)抑制劑程序性死亡受體1(PD-1)單抗在不同腫瘤中的確證性研究結(jié)果卻提示最初的ORR改善并不總能很好地預(yù)測(cè)長(zhǎng)期生存獲益[10]�����。因此��,創(chuàng)新藥如果能夠同時(shí)提供反應(yīng)替代終點(diǎn)和臨床終點(diǎn)改善程度的早期臨床研究數(shù)據(jù)并加以分析�����,是證明二者相關(guān)性的有力依據(jù)。
④ 何種限度的替代終點(diǎn)或中間臨床終點(diǎn)改善可以被認(rèn)為是“明顯改善或明顯療效”�����。一般意義的療效改善是不能支持創(chuàng)新藥物獲得附條件批準(zhǔn)的,申請(qǐng)人應(yīng)該提出合理的替代終點(diǎn)改善目標(biāo)并論證這一目標(biāo)極有可能轉(zhuǎn)化成臨床獲益��。論證可能會(huì)非常困難�����,新藥審評(píng)中不乏替代終點(diǎn)改善非?����?捎^卻未能成功轉(zhuǎn)化為長(zhǎng)期生存獲益或轉(zhuǎn)化程度遠(yuǎn)低于預(yù)期的案例��。維奈克拉聯(lián)合小劑量阿糖胞苷(LDAC)治療不適合接受強(qiáng)化治療的急性髓細(xì)胞白血?�。ˋML)患者�����,將完全緩解率(CR��,包括伴外周血細(xì)胞計(jì)數(shù)部分恢復(fù))由LDAC的15%提高到47%���,卻未獲得總生存期(OS)的改善(HR=0.75���,P=0.11)[11]��。因此���,附條件批準(zhǔn)并非只關(guān)注替代終點(diǎn)是否達(dá)到預(yù)期目標(biāo),已經(jīng)獲得的臨床終點(diǎn)數(shù)據(jù)是預(yù)測(cè)臨床獲益時(shí)非常關(guān)鍵的審評(píng)依據(jù)�����。
⑤ 確證性研究的具體要求���。附條件批準(zhǔn)是加快藥物上市的注冊(cè)路徑��,但不應(yīng)該影響臨床研發(fā)計(jì)劃的正常推進(jìn)��,更不能成為中斷或延遲開展確證性研究的理由���。申請(qǐng)人應(yīng)該在溝通附條件批準(zhǔn)上市技術(shù)要求的同時(shí),就確證性研究的具體要求與審評(píng)部門達(dá)成一致�����。按照一般的研發(fā)規(guī)律��,如果支持附條件批準(zhǔn)的臨床研究結(jié)果積極�����,申請(qǐng)人應(yīng)該盡快啟動(dòng)確證性研究�����。附條件批準(zhǔn)的藥品注冊(cè)證書將載明具體的確證性研究�����,原則上���,持有人不應(yīng)以研究進(jìn)行不順利或結(jié)果與預(yù)期不相符等原因�����,對(duì)確證性研究的目標(biāo)人群���、治療方案、對(duì)照治療���、主要療效終點(diǎn)等要素進(jìn)行實(shí)質(zhì)性變更��。
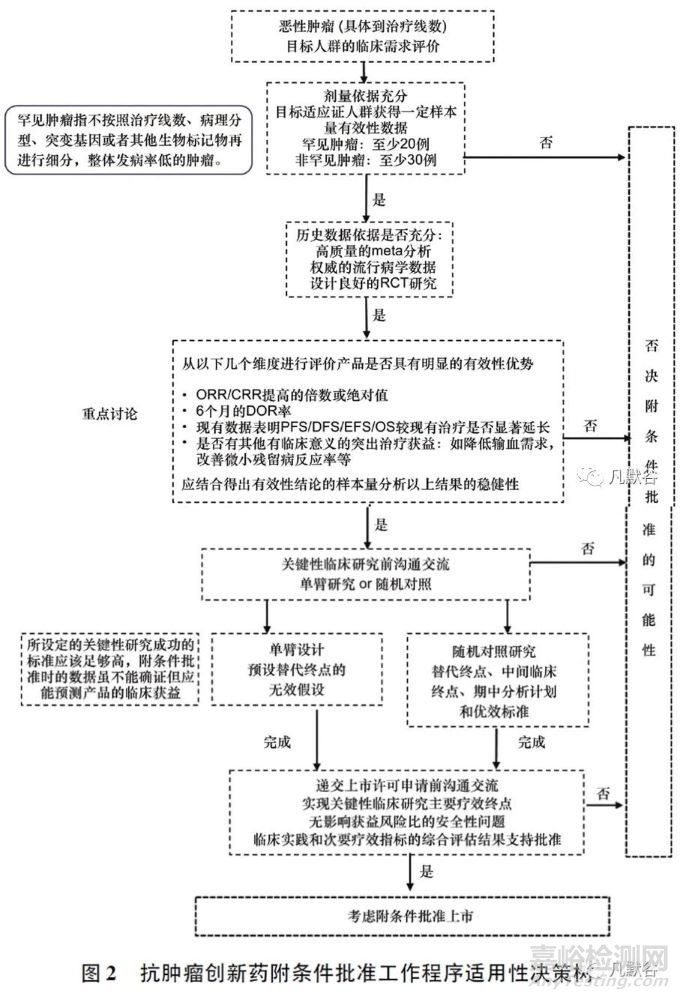
4��、附條件批準(zhǔn)適用性的決策過(guò)程
附條件批準(zhǔn)適用性決策過(guò)程中的考慮要點(diǎn)和一般標(biāo)準(zhǔn)如圖2所示�����。創(chuàng)新藥應(yīng)基于本品已獲得的臨床研究數(shù)據(jù)與審評(píng)部門展開附條件批準(zhǔn)的溝通交流�����,而不是以同類藥物的用藥經(jīng)驗(yàn)作為溝通基礎(chǔ)���。一般而言�����,至少需要在目標(biāo)適應(yīng)證人群中獲得20~30例受試者(因瘤種而異)的有效性數(shù)據(jù)后���,審評(píng)部門才會(huì)啟動(dòng)附條件批準(zhǔn)的溝通交流。在缺乏可靠的歷史數(shù)據(jù)時(shí)��,申請(qǐng)人應(yīng)考慮開展小規(guī)模的隨機(jī)對(duì)照研究作為附條件批準(zhǔn)適用性的決策依據(jù)��。
需要說(shuō)明的是,一個(gè)藥品是否可以獲得附條件批準(zhǔn)���,受到其創(chuàng)新性���、申請(qǐng)人和學(xué)術(shù)界對(duì)產(chǎn)品機(jī)制和疾病的了解程度��、目標(biāo)適應(yīng)證人群臨床需求的迫切程度等多方面因素的影響���,對(duì)所有附條件批準(zhǔn)提出一個(gè)完全量化和固化的決策標(biāo)準(zhǔn)是不切實(shí)際也有失公平的��。
由于附條件批準(zhǔn)程序的核心是以臨床價(jià)值為導(dǎo)向���,審評(píng)部門在每個(gè)溝通環(huán)節(jié)的建議均以解決目標(biāo)治療人群當(dāng)時(shí)的臨床需求為前提,藥物最終是否可獲得附條件批準(zhǔn)上市��,將取決于目標(biāo)適應(yīng)證人群在做出監(jiān)管決策時(shí)可獲得的治療手段及臨床數(shù)據(jù)所體現(xiàn)的治療優(yōu)勢(shì)�����。這意味著申請(qǐng)人在制定整體臨床研發(fā)計(jì)劃時(shí)應(yīng)具有前瞻性���,并且在臨床實(shí)踐發(fā)生重要變化時(shí)及時(shí)調(diào)整研發(fā)策略�����。
5���、附條件批準(zhǔn)工作程序?qū)嵤┻^(guò)程中的監(jiān)管問(wèn)題
對(duì)附條件批準(zhǔn)工作程序?qū)嵤┻^(guò)程發(fā)現(xiàn)的重要問(wèn)題進(jìn)行梳理并嘗試提出解決建議���。
1)藥品獲得附條件批準(zhǔn)后,對(duì)其后相同適應(yīng)證的附條件批準(zhǔn)申請(qǐng)的影響
在溝通交流階段��,同期可能有多個(gè)藥品在同一適應(yīng)證人群中獲得了相當(dāng)積極的早期研究數(shù)據(jù)�����,并同時(shí)按照附條件批準(zhǔn)策略開展臨床研究���。如果藥品A率先獲得附條件批準(zhǔn)上市�����,由于藥品A的確證性研究能否獲得成功存在不確定性���,其他藥品在完成關(guān)鍵性注冊(cè)研究后仍然可以按照溝通交流中與審評(píng)部門達(dá)成的共識(shí)遞交附條件批準(zhǔn)申請(qǐng)。
但是如果藥品A上市后完成了確證性研究并成功獲得常規(guī)批準(zhǔn)���,該適應(yīng)證人群的現(xiàn)有治療手段和臨床實(shí)踐便因此發(fā)生變化��,其附條件批準(zhǔn)的適用性或技術(shù)要求也會(huì)隨之改變�����,按照前期確定的技術(shù)要求開展研究并在藥品A獲常規(guī)批準(zhǔn)后提出的附條件批準(zhǔn)申請(qǐng)很可能會(huì)被拒絕���,而與藥品A作用機(jī)制相同的其他藥品則將不再符合附條件批準(zhǔn)該適應(yīng)證的適用情形。
2)符合附條件批準(zhǔn)的藥品與優(yōu)先審評(píng)審批程序
按照現(xiàn)行辦法[1]���,“符合附條件批準(zhǔn)的藥品”可以申請(qǐng)適用優(yōu)先審評(píng)審批程序��。原則上��,只有臨床研究數(shù)據(jù)提示明顯臨床優(yōu)勢(shì)的藥品才有可能符合附條件批準(zhǔn)��,通常會(huì)被納入優(yōu)先審評(píng)審批程序���。但是,如果已有相同作用機(jī)制的藥品被附條件批準(zhǔn)�����,后續(xù)遞交相同適應(yīng)證附條件批準(zhǔn)申請(qǐng)藥品的臨床價(jià)值將被大大降低,因此不能再獲得優(yōu)先審評(píng)審批�����。
3)附條件批準(zhǔn)藥品與“現(xiàn)有治療手段”
《藥品附條件批準(zhǔn)上市技術(shù)指導(dǎo)原則(試行)》指出“附條件批準(zhǔn)上市的藥品��,在臨床獲益未經(jīng)證實(shí)前不作為現(xiàn)有治療手段”�����。但該指導(dǎo)原則同時(shí)說(shuō)明“現(xiàn)有治療手段是指在境內(nèi)已批準(zhǔn)用于治療相同疾病的藥品���,或者標(biāo)準(zhǔn)治療方法等”���。臨床實(shí)踐對(duì)標(biāo)準(zhǔn)治療的定義與藥品獲批適應(yīng)證不完全一致的現(xiàn)象并不少見,例如: 阿扎胞苷在全球范圍內(nèi)均未被監(jiān)管機(jī)構(gòu)批準(zhǔn)用于骨髓原始細(xì)胞≥30%的AML��,卻已經(jīng)成為不能接受強(qiáng)化化療AML患者的標(biāo)準(zhǔn)治療��。
由于一些歷史原因��,一些附條件批準(zhǔn)上市的藥品未開展確證性注冊(cè)研究(如苯達(dá)莫司汀用于惰性非霍奇金淋巴瘤)��,但其臨床獲益卻在臨床實(shí)踐中得到證實(shí)且其價(jià)值得到廣泛認(rèn)可�����,甚至成為標(biāo)準(zhǔn)用藥方案的一部分,沒有理由將這些附條件批準(zhǔn)的藥品排除在現(xiàn)有治療手段之外���。即使不作為“現(xiàn)有治療手段”��,附條件批準(zhǔn)藥品的研究結(jié)果也構(gòu)成歷史數(shù)據(jù)的一部分��,為相同適應(yīng)證中其他藥品的附條件批準(zhǔn)申請(qǐng)?zhí)峁┖芎玫膶徳u(píng)參考�����。
針對(duì)類似情形��,歐盟指出: “為證明第二種或后續(xù)產(chǎn)品對(duì)未滿足臨床需求的價(jià)值時(shí),應(yīng)考慮前期已獲得條件性批準(zhǔn)藥品所累積的臨床數(shù)據(jù)和其療效尚未解決的不確定性���。由于已獲條件批準(zhǔn)的藥品尚未確認(rèn)其全部獲益�����,若預(yù)期后續(xù)產(chǎn)品可與已獲條件性批準(zhǔn)產(chǎn)品同等程度或更大程度地滿足未滿足的臨床需求�����,也可獲得條件性批準(zhǔn)的推薦��。”
4)附條件批準(zhǔn)藥品注冊(cè)證書有效期及上市后研究完成時(shí)限
現(xiàn)行辦法沒有指定附條件批準(zhǔn)藥品注冊(cè)證書的有效期和確證性研究的完成時(shí)限���,但常規(guī)批準(zhǔn)藥品的證書有效期為5年[1]��,因此附條件批準(zhǔn)藥品注冊(cè)證書的有效期不能長(zhǎng)于5年��。確證性研究數(shù)據(jù)補(bǔ)充申請(qǐng)的審評(píng)時(shí)限為200d[1]���,因此原則上需要在附條件批準(zhǔn)后4年內(nèi)遞交申請(qǐng)。
實(shí)際上��,敦促持有人在藥品附條件批準(zhǔn)上市后盡快完成確證性研究��,對(duì)所有監(jiān)管機(jī)構(gòu)而言都是一個(gè)非?����,F(xiàn)實(shí)的問(wèn)題���,設(shè)立有效期是解決方案之一��。對(duì)美國(guó)FDA加速批準(zhǔn)和歐盟條件性批準(zhǔn)的分析表明��,自獲批上市至采取后續(xù)行動(dòng)所需時(shí)間超過(guò)6年的比例分別為21%和27%[12]��,因此��,中國(guó)的時(shí)限要求將給附條件批準(zhǔn)藥品帶來(lái)不小的壓力��。
美國(guó)FDA的數(shù)據(jù)表明�����,在獲得加速批準(zhǔn)時(shí)已經(jīng)啟動(dòng)確證性研究的藥品�����,自批準(zhǔn)至完成確證所需的時(shí)間明顯縮短(3.1vs5.5年)[12]�����,一方面是因?yàn)樵鐔?dòng)直接帶來(lái)的時(shí)間優(yōu)勢(shì)�����,另一方面是因?yàn)樵谒幤帆@得加速批準(zhǔn)后再啟動(dòng)確證性研究�����,受試者對(duì)對(duì)照治療的態(tài)度會(huì)受到試驗(yàn)藥品的預(yù)期獲益影響而使入組難度增加���。
總結(jié)
自實(shí)施之日起�����,加快上市注冊(cè)程序的具體流程和決策標(biāo)準(zhǔn)即廣受關(guān)注��。審評(píng)部門作為直接的實(shí)踐者��,力爭(zhēng)公開透明��,最大程度地發(fā)揮加快上市注冊(cè)程序?qū)膭?lì)創(chuàng)新的激勵(lì)作用��,更令患者盡早受益于藥品創(chuàng)新的成果�����。
參考文獻(xiàn):
《中國(guó)新藥雜志》2023年第32卷第2期