前 言
歐盟為了能夠更有效的執(zhí)行MDR法規(guī)下規(guī)定的警戒的要求���,在2023年2月14醫(yī)療器械協(xié)調(diào)小組MDCG發(fā)布了“MDCG 2023-3關(guān)于醫(yī)療器械法規(guī)MDR(EU) 2017/745中警戒術(shù)語(yǔ)和概念的問(wèn)題和回答概述”協(xié)調(diào)文件���,該文件為了澄清關(guān)于醫(yī)療器械法規(guī)MDR(EU) 2017/745第 VII 章上市后監(jiān)管、警戒和市場(chǎng)監(jiān)管第2節(jié)警戒中概述的重要術(shù)語(yǔ)和概念��。該文件中提出的一些定義從MEDDEV 2.12-1 rev 8醫(yī)療器械警戒系統(tǒng)指南中重新引入�����,并在相關(guān)情況下進(jìn)行了修改��,以便與MDR保持一致。
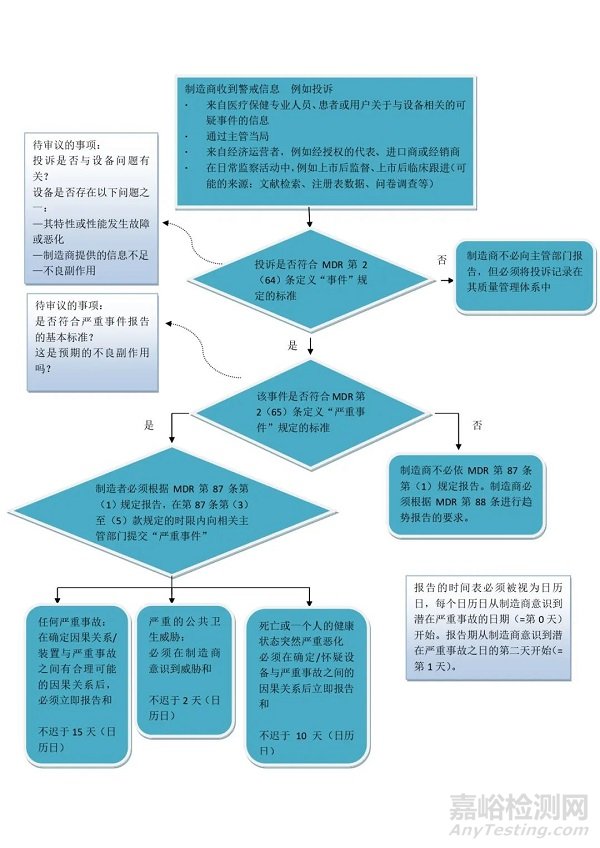
MDR法規(guī)下的警戒系統(tǒng)制造商該如何向主管部門報(bào)告事件呢���,該文件引入了MDR法規(guī)下事件和嚴(yán)重事件管理和報(bào)告遵循的流程��,以更好的來(lái)指導(dǎo)制造商如何來(lái)管理事件和嚴(yán)重事件��。
MDR法規(guī)要求
事件
是指市場(chǎng)上可獲得的器械特性或性能的任何故障或劣化事件���,包括由于人機(jī)工程學(xué)特征、制造商提供的信息中的任何不足以及任何不期望的副作用而造成的使用錯(cuò)誤���。
根據(jù)MDR要求���,事件不向主管當(dāng)局報(bào)告。但是�����,事件必須在制造商的質(zhì)量管理體系中記錄和考慮��,并根據(jù)趨勢(shì)報(bào)告的要求報(bào)告�����。
嚴(yán)重事件
是指直接或間接導(dǎo)致��、有可能導(dǎo)致或可能會(huì)導(dǎo)致以下任一狀況的任何事件:
(a) 患者���、使用者或其他人員死亡���;
(b) 患者、使用者或其他人員健康狀態(tài)的暫時(shí)性或永久性嚴(yán)重惡化�����;
(c) 嚴(yán)重公眾健康威脅�����。
具體地說(shuō)�����,嚴(yán)重事件是直接或間接導(dǎo)致��、可能導(dǎo)致或可能導(dǎo)致患者��、使用者或其他人死亡或健康狀況暫時(shí)或永久嚴(yán)重惡化或構(gòu)成嚴(yán)重公共健康威脅的事件的子集�����。
報(bào)告要求
在歐盟市場(chǎng)上提供器械的器械制造商,應(yīng)向相關(guān)主管機(jī)構(gòu)報(bào)告以下內(nèi)容:
(a)任何涉及在歐盟市場(chǎng)上銷售器械的嚴(yán)重事件�����,但不包括產(chǎn)品信息中清楚地記錄并在技術(shù)文件中量化的預(yù)期副作用外�����,并根據(jù)趨勢(shì)報(bào)告要求報(bào)告���;
(b)任何有關(guān)歐盟市場(chǎng)上銷售器械的現(xiàn)場(chǎng)安全糾正措施�����,若現(xiàn)場(chǎng)安全糾正措施的原因并不僅限于在第三類國(guó)家銷售的器械���,則包括第三國(guó)對(duì)在歐盟市場(chǎng)上合法提供的器械所采取的任何現(xiàn)場(chǎng)安全糾正措施。
制造商應(yīng)在制造商與其器械建立了事件之間因果關(guān)系后或者發(fā)現(xiàn)這種因果關(guān)系合理時(shí)�����,立即報(bào)告任何嚴(yán)重事件��,這一時(shí)限不遲于其意識(shí)到嚴(yán)重事件后的 15 天。
若出現(xiàn)嚴(yán)重的公共衛(wèi)生事件��,則應(yīng)立即報(bào)告���,且不遲于制造商察覺(jué)到此威脅后 2 天。
若出現(xiàn)人員死亡或健康狀況意外嚴(yán)重惡化���,該報(bào)告應(yīng)在制造商確認(rèn)或可疑器械與嚴(yán)重事件之間的因果關(guān)系后立即提供�����,且不遲于制造商察覺(jué)到該嚴(yán)重事件之日后 10 天��。
事件和嚴(yán)重事件管理流程