摘要
我國現(xiàn)行的藥品上市后變更管理思路借鑒了歐美上市后變更管理制度����,本研究首先介紹美國、歐盟及中國的變更管理制度����,然后重點對我國近期發(fā)布實施的《已上市化學藥品藥學變更研究技術(shù)指導原則(試行)》進行介紹,以助于更好地理解和使用該變更指導原則��。
背景
藥品上市后變更管理是藥品全生命周期管理的重要組成部分����。《藥品上市后變更管理辦法 ( 試行 )》(以下簡稱《辦法》) 是我國首部專門針對藥品上市后變更設置的規(guī)范性文件����,其實施目的和意義主要體現(xiàn)在以下兩方面 :一方面鼓勵藥品上市許可持有人 (marketing authorization holder,MAH) 運用新生產(chǎn)技術(shù)�����、新方法��、新設備����、新科技成果,不斷改進和優(yōu)化生產(chǎn)工藝��,持續(xù)提高藥品質(zhì)量�����,提升藥品安全性、有效性和質(zhì)量可控性 �����;另一方面�����,堅決貫徹習近平總書記對于藥品監(jiān)管工作“四個最嚴”的要求����,規(guī)范藥品變更行為和變更監(jiān)管,嚴厲打擊非法變更�����,落實MAH 主體責任����,保障人民群眾用藥安全。
我國的藥品上市后變更管理主要參考美國 FDA和歐盟 EMA 等先進管理理念制修訂����。本研究首先介紹美國 FDA 和歐盟 EMA 的藥品上市后變更管理制度��,然后介紹我國現(xiàn)行的變更管理制度����,最后對我國新發(fā)布實施的《已上市化學藥品藥學變更研究技術(shù)指導原則 ( 試行 )》( 以下簡稱“新版變更指導原則”) 進行簡要介紹����,以助于更好地理解和執(zhí)行�����。
一�����、美國藥品上市后變更管理簡介
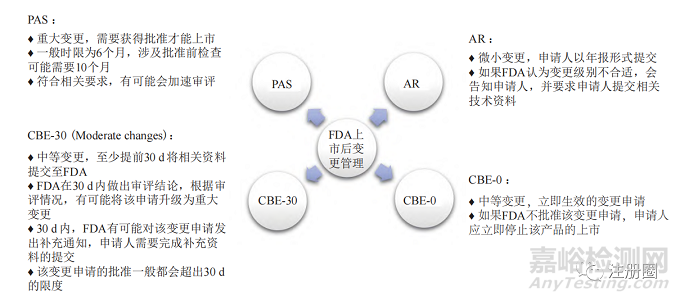
美國 FDA 依據(jù)變更對產(chǎn)品質(zhì)量產(chǎn)生影響的可能性����,將已上市產(chǎn)品的變更分為批準前補充申請 (prior approval supplement,PAS)�����、30 d 后執(zhí)行(changes being effffected in 30 days�����,CBE-30)、立即生效 (changes being effected����,CBE-0) 和年度報告(annual report,AR)4 種情形��。其變更管理概況見圖1����。
▲ 圖1-美國 FDA 上市后變更管理概況
Fig.1 Overview of Post-approval Change Management of FDA
很難在一個變更指導原則中囊括所有的變更事項,F(xiàn)DA 針對變更發(fā)布了多個指南文件 [1—5]����,針對不同劑型發(fā)布的變更指導原則包括速釋口服固體制劑、緩釋口服固體制劑及非無菌半固體制劑的擴大規(guī)模和上市后變更等����。
二、歐盟 EMA 上市后變更管理簡介
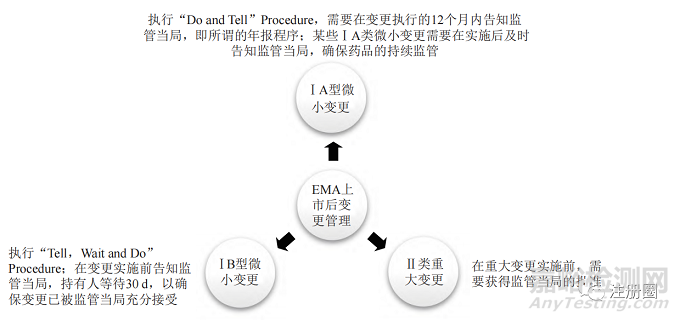
歐盟 EMA 的上市后變更管理程序與 FDA 基本相似�����,依據(jù)變更對產(chǎn)品質(zhì)量產(chǎn)生影響的可能性將變更分類劃分為Ⅰ A、Ⅰ B 和Ⅱ類變更�����。歐盟上市后變更管理程序簡介見圖2�����。
▲ 圖2-歐盟 EMA 上市后變更程序簡介
Fig.2 Post-approval Change Management Protocols of EMA
歐盟上市后變更指導原則除了規(guī)定上述ⅠA��、ⅠB 和Ⅱ類變更外����,還規(guī)定了擴展 ( extensions)和緊急安全性限制 (urgent safety restrictions) 等內(nèi)容 [6]����。前者包括規(guī)格變更、劑型變更或給藥途徑變更等 ��;后者指申請人或監(jiān)管機構(gòu)對威脅公眾健康的事件采取的即刻變更程序����,緊急安全限制可以在發(fā)起后15d 內(nèi)提交正式變更申請。
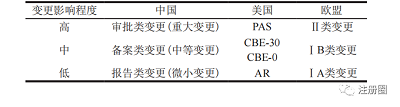
在藥品上市后變更管理方面�����,中國充分借鑒了歐美上市后變更管理思路,引入了依風險進行變更管理的理念��,同時體現(xiàn)了與國際接軌的監(jiān)管思路�����。中美歐對藥品上市后變更均是按照變更的程度和風險等級劃分管理要求�����,即根據(jù)變更對藥品安全性�����、有效性或質(zhì)量可控性產(chǎn)生影響的可能性設定不同的變更等級�����,并分別設立了相應的遞交途徑��、審批時限和變更實施原則��。詳見表 1��。
▲ 表1-中美歐變更管理情況對比
Tab.1 Comparison of Change Managements
between China, the United States and Europe
我國將變更級別分為重大變更、中等變更�����、微小變更�����。需要強調(diào)的是��,重大變更不一定會對產(chǎn)品質(zhì)量產(chǎn)生實質(zhì)性影響����,之所以將某些變更事項劃分為重大變更��,是因為該變更對產(chǎn)品質(zhì)量����、安全性及有效性等產(chǎn)生影響的可能性比較大,而微小變更對產(chǎn)品質(zhì)量��、安全性及有效性產(chǎn)生影響的可能性比較小����。
四��、“新版變更指導原則”簡介
在對新舊兩版“已上市化學藥品變更指導原則”進行對比的基礎上��,本研究對新版變更指導原則進行簡要介紹����,希望更好地促進該指導原則的貫徹執(zhí)行�����。
4.1 新舊兩版“已上市化學藥品變更指導原則”對比
隨著新的《藥品注冊管理辦法》落地實施��,之前的變更管理思路已不再符合現(xiàn)行的法規(guī)技術(shù)要求��。為了與新法規(guī)配套����,國家藥品監(jiān)督管理局藥品審評中心 (CDE) 組織修訂了“新版變更指導原則”,并正式發(fā)布實施����。為更好地理解新版變更指導原則,現(xiàn)對新舊兩版上市后變更指導原則進行簡單對比��,以突出修訂重點��,便于理解和執(zhí)行。詳見表 2����。
▲ 表2-新舊兩版已上市化學藥品變更研究的技術(shù)指導原則對比
Tab.2 Comparison of Technical Guidelines for Change Research of Listed Chemical Drugs between the Old and New Editions
部分變更分類與原來的法規(guī)指南相比有了較大改變。比如��,藥品分包裝由審批改為備案�����,很大程度上簡化了流程����、縮短了時限 ;再比如��,審批類變更納入“持有人轉(zhuǎn)讓藥品上市許可”����,這意味著MAH 的變更被視為重大變更��,MAH 的資質(zhì)將在藥品上市許可轉(zhuǎn)讓過程中被重點考察 ����;新的變更管理制度增加了報告類的變更途徑����,企業(yè)可采用年度報告的方式報告微小變更 ( 實際生產(chǎn)過程中����,發(fā)生微小變更的頻次是最高的,重大變更發(fā)生的頻次較少 )�����,無需審評審批��,提升了 MAH 實施變更的及時性和自由度�����。
4.2 新版變更指導原則的相關(guān)章節(jié)介紹
4.2.1 變更生產(chǎn)場地
MAH 制度下�����,與以前最大的變化是生產(chǎn)場地發(fā)生變更��,所以法規(guī)單獨拿出來�����,分別對 MAH 變更和生產(chǎn)場地變更制定了管理要求,此部分內(nèi)容詳見《辦法》和《藥品生產(chǎn)監(jiān)督管理辦法》�����。
僅發(fā)生 MAH 變更��,受讓方應取得相應生產(chǎn)范圍的藥品生產(chǎn)許可證�����,并向 CDE 提出補充申請����,此外變更后的 MAH 也需要進行 GMP 符合性檢查,確保變更后的 MAH 能夠履行責任����,并能夠持續(xù)穩(wěn)定地生產(chǎn)出符合質(zhì)量要求的產(chǎn)品。
生產(chǎn)場地變更�����,包括生產(chǎn)地址的改變或新增����,或同一生產(chǎn)地址內(nèi)的生產(chǎn)場地的新建、改建��、擴建����。關(guān)于生產(chǎn)場地變更,新版變更指導原則中沒有按照其他注冊事項變更進行微小����、中等及重大變更情況的描述,而是指出“生產(chǎn)場地變更需要按照《藥品生產(chǎn)監(jiān)督管理辦法》及《藥品上市后變更管理辦法( 試行 )》相關(guān)規(guī)定執(zhí)行”��。生產(chǎn)場地變更一般由監(jiān)管經(jīng)驗比較豐富的省局進行管理����。
2008 年的變更指導原則中,國產(chǎn)藥無變更生產(chǎn)場地的相關(guān)規(guī)定����,原因是之前的藥品批準文號直接與生產(chǎn)企業(yè)關(guān)聯(lián),如果生產(chǎn)企業(yè)發(fā)生變更��,只能走技術(shù)轉(zhuǎn)移的申報路徑�����,先將自己的批準文號注銷才能完成產(chǎn)品從一個生產(chǎn)企業(yè)轉(zhuǎn)移至另外一個生產(chǎn)企業(yè)。因 MAH 制度的實施�����,藥品批準文號不再直接與生產(chǎn)企業(yè)關(guān)聯(lián)����,而是隸屬于 MAH,所以新版變更指導原則增設變更生產(chǎn)場地的相關(guān)規(guī)定����。新版變更指導原則雖未對生產(chǎn)場地變更進行變更等級劃分,但對生產(chǎn)場地變更的研究驗證工作進行了規(guī)定�����,MAH/ 生產(chǎn)企業(yè)在申報原料藥或制劑的生產(chǎn)場地變更時可以參考進行研究驗證工作����。
4.2.2 變更制劑處方中的輔料
制劑處方中的輔料變更情形主要包括變更輔料的種類、用量�����、技術(shù)等級、供應商�����、質(zhì)量標準等����,新版變更指導原則細化了普通口服固體制劑�����、口服緩釋 / 控釋制劑�����、腸溶制劑及非無菌半固體制劑這3 大類劑型的變更情況及驗證研究工作��。指導原則中未涵蓋的劑型一般比較復雜或是輔料變更風險比較高����,故對指導原則中未涵蓋劑型的輔料用量變更按照重大變更進行嚴格管理。由于輔料對治療窗窄及生物藥劑學分類系統(tǒng) (BCS) Ⅳ類的藥物影響較顯著����,故當窄窗藥物及 BCS Ⅳ類藥物的輔料用量變化超出微小變更范圍時,均應按照重大變更進行研究。
與舊版變更指導原則相比����,新版變更指導原則的變化較大之處在于將非無菌半固體制劑中防腐劑用量變更單獨進行規(guī)定。具體允許的防腐劑變更幅度分別規(guī)定為 :微小變更——變更不超過原批準用量的 10% ����;中等變更——變更大于原批準用量的10%、不超過 20% ��;重大變更——變更超過原批準用量的20% (包括刪除防腐劑 )或變更防腐劑種類��。變更防腐劑應提交的研究驗證工作與其他變更情況相比有一個不同之處�����,即不管變更分類是微小變更還是重大變更��,都應提供規(guī)定范圍內(nèi)抑菌劑最低濃度的抑菌效力試驗�����,以證明變更后產(chǎn)品的微生物限度符合要求����。
由于不同劑型的不同變更類別涵蓋的變更事項在指導原則中描述得較為詳細��,故本研究不再贅述����。
4.2.3 變更生產(chǎn)批量
變更生產(chǎn)批量為新版變更指導原則中的新增章節(jié)��,包括原料藥批量和制劑批量變更�����。原料藥的批量變更只有微小和中等兩個變更分類��,制劑的批量變更依風險分為微小�����、中等和重大變更��。
不同的劑型或不同的制備工藝��,其批量變更風險不同����,制劑批量變更中的重大變更一般為特殊劑型制劑 ( 如復雜工藝的緩控釋制劑及腸溶制劑�����、透皮給藥制劑、脂質(zhì)體����、長效制劑等 ) 的生產(chǎn)批量變更。這里需要關(guān)注的是緩控釋制劑及腸溶制劑的修飾詞“復雜工藝”�����,例如工藝較為簡單的骨架型緩釋片�����,其批量變更對其質(zhì)量產(chǎn)生影響的可能比較小����,此時可以降低其變更分類。
批量變更往往伴隨著生產(chǎn)設備和 ( 或 ) 工藝參數(shù)的變更��,所以批量變更的所有變更分類��,不論是重大變更還是微小變更均需進行工藝驗證�����,提供批生產(chǎn)記錄,并進行質(zhì)量對比研究����,對樣品批次的要求也有所增加。
4.2.4 變更制劑所用原料藥的供應商
變更制劑所用原料藥的供應商不應對藥品安全性����、有效性和質(zhì)量可控性產(chǎn)生負面影響。舊版變更指導原則將原料藥變更劃分為進口和國產(chǎn)�����,分別進行變更情況的描述����,而新版變更指導原則不再區(qū)分進口和國產(chǎn)產(chǎn)品��,統(tǒng)一要求�����。
變更制劑所用原料藥的供應商一般按照中等變更進行管理����,變更后的原料藥如尚未獲得批準����,則按照重大變更管理����。不管是中等變更還是重大變更均需要進行較為詳細的研究驗證工作,以證明變更制劑所用原料藥的供應商不會對藥品安全性��、有效性和質(zhì)量可控性產(chǎn)生負面影響��。
4.2.5 變更注冊標準��、有效期和貯存條件
變更藥品注冊標準的適用范圍一般包括變更原料藥及制劑注冊標準中的檢驗項目�����、檢驗方法��、限度等����。注冊標準是反映和控制藥品質(zhì)量的重要手段,與上市后藥品抽檢及其質(zhì)量保障密切相關(guān)�����,為方便上市后監(jiān)管,將注冊標準變更風險等級劃分為中等和重大�����,沒有微小變更的分類����。
藥品有效期和貯存條件變更可能包含以下幾種情況 :①延長或縮短有效期 ;②嚴格或放寬貯存條件��。變更可能只涉及上述某一種情況的變更��,也可能涉及上述多種情況的變更�����。一般情況下�����,有效期的變更為中等變更��,而貯存條件變更均為重大變更��,若貯存條件發(fā)生變更��,應提供充分的變更研究資料��。
4.2.6 變更原料藥和制劑生產(chǎn)工藝
變更原料藥生產(chǎn)工藝主要指化學合成原料藥生產(chǎn)工藝或半合成原料藥的化學合成及之后生產(chǎn)工藝的變更����,主要包括合成路線、生產(chǎn)條件����、物料控制 / 過程控制及其他可能的變更。此部分內(nèi)容充分體現(xiàn)了與國際接軌的監(jiān)管思路����,其先進性及科學性主要體現(xiàn)在以下兩方面 :①變更合成路線的,變更后合成路線中起始物料的選擇應符合人用藥品技術(shù)要求國際協(xié)調(diào)理事會 (ICH)Q11 的相關(guān)要求 ����;②在對比研究中,雜質(zhì)譜一致的認定條件體現(xiàn)了與 ICHQ3A�����、ICH Q3C����、ICH Q3D 及 ICH M7 的接軌��。
通過風險評估來確定原料藥的變更分類����,非無菌原料藥的變更分類依據(jù)變更對原料藥雜質(zhì)譜以及關(guān)鍵質(zhì)量屬性產(chǎn)生影響的程度來劃分�����,而無菌原料藥還需要評估變更對其無菌保障水平產(chǎn)生的影響����。風險評估中,變更越靠近成品����,風險越高 ;變更合成路線����,或合成工藝中使用新的試劑 / 溶劑����,其雜質(zhì)譜產(chǎn)生變化的風險較高。按照確定的變更分類提供相應的研究驗證資料。
制劑生產(chǎn)工藝變更主要包括變更制劑生產(chǎn)過程及工藝參數(shù)����、變更原料藥內(nèi)控標準 / 制劑中間體內(nèi)控標準或生產(chǎn)過程控制、變更制劑生產(chǎn)設備����、變更制劑的外形等。以變更制劑外形為例�����,其變更風險依據(jù)變更對制劑的影響程度以及制劑劑型和制劑生產(chǎn)工藝的復雜程度等確定�����。普通口服片劑����、膠囊劑或栓劑形狀、尺寸的微小變化歸類為微小變更����,而發(fā)生顯著變化的為中等變更,但不管是形狀����、尺寸的微小變更還是中等變更����,變更前后的溶出行為均不能發(fā)生改變 ����;變更緩控釋制劑的形狀、尺寸和刻印或者增加 ( 或刪除 ) 片劑的功能性刻痕均為重大變更�����,對產(chǎn)品關(guān)鍵質(zhì)量屬性產(chǎn)生影響的風險較大��,需提供充分的研究驗證工作�����。
制劑生產(chǎn)工藝變更情形復雜�����,變更指導原則不能涵蓋可能的所有變更情形�����,MAH 應當根據(jù)內(nèi)部變更分類原則����、工作程序和風險管理標準,結(jié)合產(chǎn)品特點����,參考有關(guān)技術(shù)指導原則,在充分研究����、評估和必要驗證的基礎上確定變更管理類別。
4.2.7 變更包裝材料和容器
包裝材料和容器是藥品的組成部分�����,主要指直接接觸藥品的包裝材料和容器��。包裝材料和容器的變更包括改變����、增加或去除的情形,以上變更情形可能對藥品的理化性質(zhì)��、雜質(zhì)譜��、含量、穩(wěn)定性�����、無菌保障水平等產(chǎn)生影響��。包裝材料和容器的變更依風險分為微小�����、中等和重大變更��,此部分變更應重點關(guān)注高風險制劑 ( 比如眼用制劑或注射劑 ) 的包材變更����,同時還應關(guān)注全新材料、全新結(jié)構(gòu)����、風險度提高的新用途的包裝材料和容器的使用,對產(chǎn)品質(zhì)量產(chǎn)生風險的可能性較高��。
對于重大變更的研究驗證工作可能需要進行包材相容性研究��,對于密封件的變更還需開展包裝密封性研究等�����。
4.2.8 新增規(guī)格
新增規(guī)格均按重大變更管理����。
新版變更指導原則和《已上市化學藥品和生物制品臨床變更技術(shù)指導原則》中均有規(guī)格變更的相關(guān)規(guī)定,這 2 個變更指導原則中規(guī)定的規(guī)格變更有本質(zhì)不同��。
藥學變更中增加規(guī)格��,是指新增規(guī)格應為原研藥品增加的新規(guī)格�����,或仿制藥增加目前原研藥品 /參比制劑已有的規(guī)格��,同時不得改變藥品原批準的適應證�����、用法用量或適用人群等�����。變更分類為重大變更�����,同時需參照現(xiàn)行的相關(guān)技術(shù)指導原則,考慮新增規(guī)格是否需要進行生物等效性研究�����。
臨床變更中增加規(guī)格��,是指新增規(guī)格的藥物含量未在已批準說明書的用法用量范圍內(nèi)�����,增加新規(guī)格通常在用法用量變更的同時提出����。此類變更為重大 A 類變更,因新增規(guī)格與尚未批準的用法用量密切相關(guān)��,需要臨床試驗數(shù)據(jù)的支持��。故此類變更一般需要提出 2 次申請����,申請人應首先提出開展支持新增規(guī)格和用法用量變更的臨床試驗申請以獲得臨床試驗批件,待臨床試驗完成����,獲得充分支持性證據(jù)后����,再次提出相應的變更申請��。
此外��,新增規(guī)格還包括多劑量包裝變更為單劑量包裝�����,比如多劑量的滴眼液變更為處方中去除防腐劑的單劑量包裝等�����,也屬于重大變更��,需進行較為詳細的研究驗證工作����。
4.2.9 關(guān)聯(lián)變更
藥品某一項變更往往不是獨立發(fā)生的��。例如��,批量變更往往同時伴隨生產(chǎn)設備及生產(chǎn)工藝的變更,處方變更可能伴隨或引發(fā)藥品注冊標準的變更����,增加規(guī)格可能會調(diào)整處方等。我們將一項變更伴隨或引發(fā)的其他變更稱之為關(guān)聯(lián)變更����。
對于關(guān)聯(lián)變更,研究工作可按照新版變更指導原則中各項變更研究工作的基本思路分別進行��,也可綜合考慮各項變更研究工作的要求而一并進行��。由于這些變更對藥品安全性��、有效性和質(zhì)量可控性產(chǎn)生的影響程度可能不同��,即這些變更可能歸屬于新版變更指導原則中各項變更的不同類別����,在按照不同類別變更相應技術(shù)要求開展研究工作時,研究工作總體上應按照技術(shù)要求較高的變更類別進行�����,同時建議關(guān)注多項關(guān)聯(lián)變更對藥品安全性、有效性和質(zhì)量可控性產(chǎn)生的疊加影響��。
五�����、結(jié)語
目前����,我國的變更管理理念主要參考了美國FDA 及歐盟 EMA 的變更管理制度,引入了依風險進行變更管理的思路����,體現(xiàn)了逐漸與國際接軌的監(jiān)管理念��,使我國的上市后變更管理更加科學����。本研究對 FDA 和 EMA 的變更管理制度進行簡單介紹,然后引入我國現(xiàn)行的變更管理制度�����,最后對我國近期發(fā)布實施的新版變更指導原則進行介紹����,促進該指導原則的正確理解和執(zhí)行�����。鼓勵 MAH 在對產(chǎn)品及其工藝��、質(zhì)量控制等不斷深入理解的基礎上����,采用 ICH 指導原則 ( 如 ICHQ12 等 ) 中的各種變更管理工具����,對變更進行研究和分類,這將有利于主動對已上市藥品進行持續(xù)改進和創(chuàng)新��。新版變更指導原則也將會在使用中不斷更新和完善��。
參考文獻
[1] FDA.Changes to an approved NDA or ANDA: guidance for industry [EB/OL].(2004-08-04)[2021-09-13].https://www.fda.gov/media/71846/download.
[2] FDA.SUPAC-IR: immediate-release solid oral dosage forms: scale-up and post-approval changes: chemistry,manufacturing and controls, in vitro dissolution testing, and in vivo bioequivalence documentation [EB/OL].(1995-11-01)[2021-09-13].https://www.fda.gov/media/70949/download.
[3] FDA.SUPAC-IR questions and answers about SUPACIR guidance [EB/OL].(1997-02-18)[2021-09-13].
https://www.fda.gov/regulatory-information/search-fdaguidance-documents.
[4] FDA.SUPAC-MR: modified release solid oral dosageforms scale-up and postapproval changes: chemistry,manufacturing, and controls; in vitro dissolution testing and in vivo bioequivalence documentation: guidance for industry [EB/OL].(1997-10-06)[2021-09-13].https://www.fda.gov/media/70956/download.
[5] FDA.SUPAC-SS: nonsterile semisolid dosage forms; scaleup and post-approval changes: chemistry, manufacturing and controls; in vitro release testing and in vivo bioequivalence documentation: guidance for industry [EB/OL].(1997-05-01)[2021-09-13].https://www.fda.gov/media/71141/download.
[6] EMA.Guidelines on the details of the various categories of variations [EB/OL].(2013-08-02)[2021-09-13].https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:C: 2013:223:FULL:EN:PDF.
[7] 國家藥品監(jiān)督管理局.已上市化學藥品變更研究的技術(shù)指導原則(一)(國食藥監(jiān)注[2008]242號)[EB/OL].[2021-09-13].https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20080515120001217.html.