某注射劑一致性評價CDE發(fā)補中指出“請結(jié)合多批次自制樣品和參比制劑的檢測結(jié)果,合理擬定有關(guān)物質(zhì)檢查項下的限度要求”�。個人認為注射劑有關(guān)物質(zhì)限度制定應(yīng)考慮自制品及參比配伍實驗的測定結(jié)果,不能僅考慮樣品穩(wěn)定期間的測定結(jié)果�,本文從國內(nèi)外的法規(guī)文件要求及案例分析兩方面入手進行闡述�,不足或錯誤之處望同行批評指正。
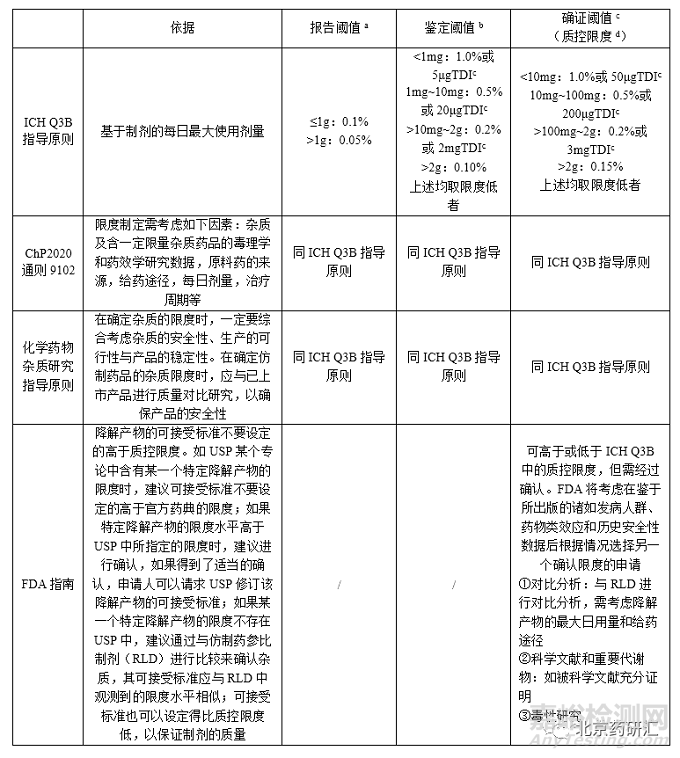
一����、現(xiàn)行法規(guī)文件中涉及有關(guān)物質(zhì)限度制定的文件主要有:ICH Q3B����、《中國藥典》2020年版通則9102“藥品雜質(zhì)分析指導(dǎo)原則”�、化學藥物雜質(zhì)研究的技術(shù)指導(dǎo)原則��、FDA指南“仿制藥(原料藥)中的雜質(zhì)研究”等��。上述文件對制劑中雜質(zhì)的限度制定對比如下�。
表1各國文件對制劑中的雜質(zhì)限度制定對比
注:a報告閾值是指超出此閾值的雜質(zhì)均應(yīng)在檢測報告中報告具體的檢測數(shù)據(jù)�。
b鑒定閾值是指超出此閾值的雜質(zhì)均應(yīng)進行定性分析����,確定其化學結(jié)構(gòu)��。
c確證閾值是指超出此閾值的雜質(zhì)均應(yīng)基于其生物安全性評估數(shù)據(jù)�,確定控制限度。
質(zhì)控限度是指質(zhì)量標準中一般允許的雜質(zhì)限度����,如制定的限度高于此限度����,則應(yīng)有充分的依據(jù)�。
TDI是指藥品雜質(zhì)的每日總攝入量��。
ICHQ3B主要是指針對創(chuàng)新藥雜質(zhì)限度制定提供了依據(jù)�,ChP2020通則9102 《藥品雜質(zhì)分析指導(dǎo)原則》�、《化學藥物雜質(zhì)研究指導(dǎo)原則》和FDA指南《仿制藥中的雜質(zhì)研究》為仿制藥雜質(zhì)限度制定提供了路徑����,但具體到注射劑類,并沒有明確規(guī)定�。
二�、案例解析
注射用阿莫西林鈉克拉維酸鉀(1.2g)
該品種在中國藥典和英國藥典中均有收載��,各國藥典標準中的有關(guān)物質(zhì)限度對比如下�。
表 2 注射用阿莫西林鈉克拉維酸鉀各國藥典標準雜質(zhì)限度對比
注:本品最大日服用劑量為阿莫西林鈉3g����。
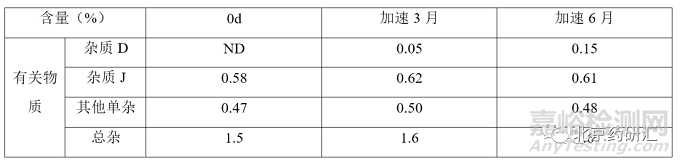
各國藥典標準中單雜最嚴限度為不得過2.5%,雜質(zhì)D和雜質(zhì)J限度均為不得過5%�,總雜最嚴為不得過7.0%;該品種加速穩(wěn)定性期間數(shù)據(jù)如下:注:本品最大日服用劑量為阿莫西林鈉3g�。
表 3 注射用阿莫西林鈉克拉維酸鉀參比制劑穩(wěn)定性數(shù)據(jù)
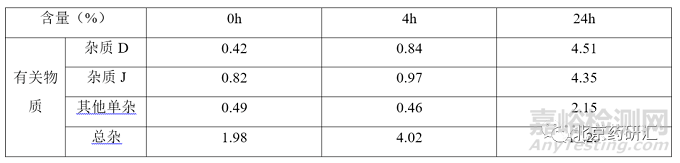
根據(jù)參比制劑穩(wěn)定性數(shù)據(jù)可知�,雜質(zhì)含量與限度要求相差甚遠,該品種效期末配伍穩(wěn)定性數(shù)據(jù)如下表所示
表 4 注射用阿莫西林鈉克拉維酸鉀參比制劑配伍穩(wěn)定性數(shù)據(jù)
由表3可知�,注射用阿莫西林鈉克拉維酸鉀各國藥典中雜質(zhì)限度的制定應(yīng)該是考慮到臨床配伍的實際應(yīng)用����。
三����、討論
通常注射劑質(zhì)量研究中會提前考察原輔料相容性��、影響因素�、加速等��,但配伍研究卻比較滯后��,往往是申報前才完成��,一但出現(xiàn)配伍研究比參比差的情況����,后果將不堪設(shè)想。因此建議在注射劑小試研究階段就開展參比及自制品的配伍實驗�,重點關(guān)注其降解雜質(zhì)�,并對其機理進行探究,確保注射劑有關(guān)物質(zhì)限度制定的合理性及臨床用藥的安全性�。
四�、參考文獻
[1] ICH Q3指導(dǎo)原則
[2] ChP2020版通則9102“藥品雜質(zhì)分析指導(dǎo)原則””
[3] CDE《化學藥物雜質(zhì)研究指導(dǎo)原則》
[4] FDA工業(yè)指南-簡化新藥申請:仿制藥中的雜質(zhì)研究