摘 要 / Abstract
目的:為我國藥品上市許可持有人制度下的偏差處理和變更控制提出建議與對策�。方法:結合我國藥品行業(yè)質量管理現(xiàn)狀,對偏差和變更進行風險分析��,提出相應的處理和控制策略�。結果:在我國實施藥品上市許可持有人制度的現(xiàn)狀下,明確了上市許可持有人與受托生產(chǎn)企業(yè)對偏差處理和變更控制的責任劃分�。結論:藥品上市許可持有人和受托生產(chǎn)企業(yè)應以風險管理為原則,采取有效措施對偏差和變更進行處理和控制��,提升藥品質量管控能力����。
Objective: To put forward suggestions and countermeasures for the deviation handling and change control under the marketing authorization holder system. Methods: Based on the current status of quality management in China’s pharmaceutical industry, risk analysis was carried out on deviations and changes, and the corresponding handling and control countermeasures were proposed. Results: Under the current marketing authorization holder system in China, responsibility divisions on deviation handling and change control between authorization holders and contract manufacturers were clarified.Conclusion: Marketing authorization holders and contract manufacturers should take effective measures to handle deviations and control changes according to the risk management principle.
藥品上市許可持有人(MAH)制度采用的是上市許可與生產(chǎn)許可分離的管理模式。我國的MAH制度自實施以來��,解除了藥品注冊和生產(chǎn)許可的捆綁模式��,極大地激發(fā)了研發(fā)機構的積極性����,使得醫(yī)藥行業(yè)創(chuàng)新活力得到有力釋放�,并且在專業(yè)分工����、資源配置、重復建設����、監(jiān)管效能上都有很大的改善和提升。MAH制度改變了藥品管理和責任承擔的機制�,MAH將承擔藥品整個生命周期內質量與風險的主要責任,這對MAH的責任和管理能力提出了更高的要求�,同時給藥品監(jiān)管帶來了新的挑戰(zhàn)。MAH制度下����,上市許可持有人和生產(chǎn)許可持有人可以是同一主體,也可以是兩個相互獨立的主體�,其允許MAH自行生產(chǎn)藥品,也可以委托其他生產(chǎn)企業(yè)進行生產(chǎn)��。在委托其他生產(chǎn)企業(yè)生產(chǎn)藥品時�,MAH如何履行責任并切實做好風險管理工作成為現(xiàn)實難題。作為《藥品生產(chǎn)質量管理規(guī)范》(GMP)的重要組成部分�,良好的偏差處理和變更控制是確保藥品質量的重要措施之一。本文通過分析國內外MAH管理以及監(jiān)管部門對偏差處理和變更控制監(jiān)管現(xiàn)狀����,為我國MAH制度下的偏差處理和變更控制提出建議與對策�,為監(jiān)管部門提出監(jiān)管政策建議����,同時為MAH加強質量風險管控體系建設����、進一步提升藥品質量管控能力提供參考。
1��、國內外MAH管理現(xiàn)狀
1.1 美國
美國是施行MAH制度最早的國家之一��?���!堵?lián)邦食品藥品和化妝品法案》(Federal Food,Drug and Cosmetic Act,F(xiàn)D&C Act)使用申請人(applicant)和申請持有人(applicant holder)來表示藥品上市許可申請人(MAA)和藥品上市許可持有人(MAH)[1-2]����。申請人和申請持有人均是藥品申請或者產(chǎn)品上市的責任主體,并且規(guī)定企業(yè)負責人是質量第一責任人�。
1.2 歐盟
歐洲經(jīng)濟共同體在其1965年頒布的Directive 65/65/EEC[3]指令提出實施MAH制度,規(guī)定藥品批準證明文件的所有者(MAH)要承擔藥品全生命周期的管理主體責任��。歐盟現(xiàn)行的MAH制度既考慮了整體性,又兼顧了成員國的自主性��。根據(jù)歐盟各國實際狀況����,可分為藥品上市許可申請人(MAA)、藥品上市許可持有人(MAH)和藥品生產(chǎn)許可持有人(PLH)三種責任主體�,分別承擔在藥品批準上市前、批準上市后批文持有者����、藥品生產(chǎn)或受托生產(chǎn)實體的三種責任,現(xiàn)行的Directive 2001/83/EC[4]沿用了MAA和MAH的定義�。
1.3 日本
2005年日本修訂生效的《藥事法》[5]正式引入MAH制度,與歐盟和美國相比較晚��,卻也是亞洲較早實施MAH制度的國家�。《藥事法》將生產(chǎn)許可和上市許可申請相互獨立����,與美國和歐盟不同的是日本提出了“上市許可人執(zhí)照”制度,必須取得某一類MAH執(zhí)照后����,才可以提出具體產(chǎn)品的上市申請����,屬于資格準入型MAH制度��。
1.4 中國
我國的MAH制度試點在2015年拉開帷幕��,2019年新修訂《藥品管理法》正式實施����,標志著我國上市許可持有人制度全面落地����。后續(xù)出臺的《藥品生產(chǎn)監(jiān)督管理辦法》[6]《藥品委托生產(chǎn)質量協(xié)議指南(2020年版)》[7]等法規(guī)的相關內容都是對MAH制度實施的補充與指導。
我國MAH制度的實施面臨著如何確保藥品從研發(fā)����、生產(chǎn)、銷售直至產(chǎn)品退市全生命周期的質量安全的挑戰(zhàn)��。同歐盟��、美國��、日本等國家和地區(qū)相比����,我國MAH制度實施較晚����,行業(yè)積累的經(jīng)驗相對不足�,跨境持有和跨境委托生產(chǎn)、境內境外主體變更相互轉換尚未完全實現(xiàn)�。基于風險考慮�,特殊藥品的委托生產(chǎn)也未完全放開,還不允許生物制品原液����、制劑分段委托生產(chǎn)。此外��,我國的藥物警戒工作也剛剛起步��,產(chǎn)品上市后安全性研究�、患者賠償保障能力相對不足,MAH質量管理能力��、風險防控能力��、責任賠償能力這“三大能力”還需要不斷提高。
2��、國內外監(jiān)管部門對偏差處理和變更控制的監(jiān)管現(xiàn)狀
2.1 美國
美國食品藥品監(jiān)督管理局(FDA)發(fā)布的多部法規(guī)����、行業(yè)指南,對MAH委托生產(chǎn)情形��,MAH和受托方的總體責任進行了界定��;推薦簽訂質量協(xié)議約定好各方的職責�,以確保委托生產(chǎn)cGMP(現(xiàn)行藥品生產(chǎn)管理規(guī)范)的符合性。美國衛(wèi)生和公共服務部(HHS)和FDA共同制定的行業(yè)指南《藥品委托生產(chǎn)質量協(xié)議》(Contract Manufacturing Arrangements for Drugs:Quality Agreements)[8]規(guī)定��,質量協(xié)議應描述MAH與受托方在藥品生產(chǎn)中各自的cGMP相關義務��、職責和活動��。解釋受托方如何向持有人報告生產(chǎn)偏差�,以及如何按照cGMP要求調查�、記錄和解決偏差;MAH和受托方均可以啟動對工藝����、設備、檢驗方法、質量標準和其他合同約定要求的變更�。雙方應對變更進行討論,在質量協(xié)議中對其進行說明����,還應列出所有變更要如何管理,包括在執(zhí)行變更之前實施所需驗證活動的職責分配�。但是對于哪些變更需要由MAH在實施之前進行審核和批準,哪些變更受托方可以直接實施����,《藥品委托生產(chǎn)質量協(xié)議》中還未給出更明確的指導意見。
2.2 歐盟
歐盟與MAH制度相關的法規(guī)主要為歐洲藥品管理局(EMA)發(fā)布的《外包服務合同框架草案》[9](Draft framework service contract for outsourcing)及《外包服務的規(guī)范流程》[10](Steps involved in outsourcing of services)等�,為歐盟藥品外包服務從法規(guī)層面提供了一個嚴謹、完整的合同樣本����,其涵蓋從合同建立到合同執(zhí)行再到合同終止的所有環(huán)節(jié),覆蓋整個藥品委托加工周期�,規(guī)范委托雙方的行為,保證各方利益��,使委托加工更具操作性��。但委托生產(chǎn)相關規(guī)定中并未提及當發(fā)生變更和偏差時�,哪些情形下由持有人批準關閉,哪些情形下由受托方批準關閉等指導意見。
2.3 日本
日本與MAH相關的文件主要為《制造委托合同》和《關于委托生產(chǎn)合同簽訂的一些注意事項》����,其中《制造委托合同》屬于一個完整的委托生產(chǎn)的合同模板,包含了簽約的目的����、制造說明書、原材料等的供應��、產(chǎn)品的生產(chǎn)量����、分包、業(yè)務執(zhí)行中的合作義務�、成品檢驗、質量保證��、提供材料的義務��、支付��、成本�、產(chǎn)品運輸����、合同解除�、無法制造��、保密����、合同期限、商談事項等17項內容�。其中涉及的變更主要是指合同本身內容發(fā)生變化,例如生產(chǎn)數(shù)量�、合同存續(xù)終止等發(fā)生變化,但對于委托生產(chǎn)中變更和偏差等內容并未進行明確闡述�。
《關于委托生產(chǎn)合同簽訂的一些注意事項》主要闡述委托生產(chǎn)前,委托方和受托方簽訂合同中應明確的事項��,文件中規(guī)定對于委托生產(chǎn)后出現(xiàn)的變更和偏差需要雙方協(xié)商處理�。
2.4 中國
我國2020年發(fā)布的《藥品委托生產(chǎn)質量協(xié)議指南(2020年版)》強調MAH的主體責任,要求偏差和變更均應由MAH審核����。MAH作為責任主體,要按照藥品監(jiān)督管理部門的規(guī)定�,全面評估、驗證變更事項對藥品安全性�、有效性和質量可控性的影響����。MAH和受托方應按照相關法律法規(guī)�、技術規(guī)范等開展變更。任何一方進行可能影響藥品質量的變更都應及時書面告知對方��。質量協(xié)議應規(guī)定雙方均須建立變更控制程序��,明確發(fā)生變更時的工作措施��;還應規(guī)定委托生產(chǎn)產(chǎn)品相關變更的風險程度由MAH評估確定�,受托方在變更實施前需要經(jīng)MAH審核批準。此外����,質量協(xié)議應規(guī)定受托方在生產(chǎn)質量管理活動中發(fā)生偏差時應按照偏差處理程序進行處理。受托方需評估與受托生產(chǎn)產(chǎn)品相關的所有偏差對產(chǎn)品安全性����、有效性和質量可控性的影響,并根據(jù)偏差的性質�、范圍及對產(chǎn)品質量的影響程度實施分類管理��,將擬采取的糾正預防措施報告MAH�。偏差處理報告需經(jīng)MAH審核批準����。
2.5 小結
綜上�,通過比較國內外制定的外包、MAH委托生產(chǎn)的流程����、框架、質量協(xié)議等相關指導原則��,均明確了MAH和受托方都應該建立各自的義務和責任�,但針對變更和偏差的具體事項,其他國家和地區(qū)的監(jiān)管機構多持開放態(tài)度�,未對雙方的責任進行界定。我國則是相對強調MAH的主體責任��,但實際在MAH制度實施過程中����,委托生產(chǎn)過程中的變更和偏差情形較為復雜,雙方對各自的責任需要更明確��、更具體的指引�。
3、我國MAH和受托生產(chǎn)企業(yè)對偏差處理和變更控制的工作程序及策略
結合MAH制度運行情況�,本文針對我國不同機構�、不同崗位人員進行問卷調查��,共收集有效問卷131份�。調查問卷按不同機構劃分,監(jiān)管機構/高校/事業(yè)單位共占7.63%����,自行生產(chǎn)的藥品上市許可持有人企業(yè)(持藥品生產(chǎn)許可證A證)占61.83%,委托生產(chǎn)的藥品上市許可持有人企業(yè)(持藥品生產(chǎn)許可證B證)占48.85%�,接受藥品上市許可持有人委托的企業(yè)(持藥品生產(chǎn)許可證C證)占21.37%。
3.1 變更控制
變更問題主要涵蓋機構與人員����、廠房與設施、設備�、物料與成品、文件管理����、生產(chǎn)管理、質量控制與質量保證��、委托生產(chǎn)與委托檢驗等八方面內容�,接受問卷調查者需選擇每個變更問題由受托方?jīng)Q定、持有人決定還是由雙方共同決定�。根據(jù)調查結果顯示受托方對于其人員管理、車間資產(chǎn)配置有較大的主動權��;機構與人員����、廠房設施、設備相關的變更傾向于由受托方?jīng)Q定或由雙方共同決定����;物料與成品管理更傾向于由MAH決定,其他的變更事項支持雙方共同決定����,調查結果詳見表1。
3.2 偏差處理
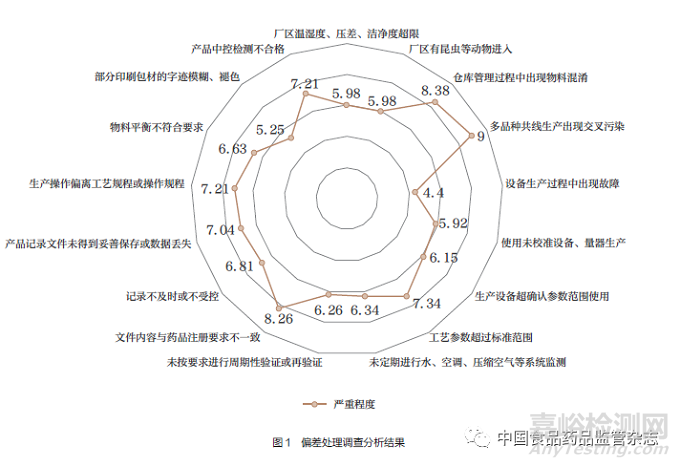
偏差問卷調查中對應其嚴重程度分為4檔����,1~3分為一般,4~5分為較嚴重��,6~8分為嚴重����,9~10分為極為嚴重。接受調查者需要給所有偏差問題進行打分�,并且還需要給出幾分以下可由受托生產(chǎn)企業(yè)自行關閉的建議��。
本文偏差共設置17項問題��,收集到的嚴重程度評分均為4分以上�,結果詳見圖1�。
問卷結果支持4分以下的偏差可由受托生產(chǎn)企業(yè)自行關閉,即大多數(shù)從業(yè)人員接受一般偏差由受托方自行關閉��,較嚴重�、嚴重、極為嚴重的偏差均需匯報給MAH�。由此可知從業(yè)人員對于偏差的處置較為謹慎。
3.3 監(jiān)管主體
根據(jù)監(jiān)管職責的調查結果顯示:對于偏差處理的監(jiān)管�,接受調查者認為應由受托生產(chǎn)企業(yè)所在地藥品監(jiān)管部門監(jiān)管的占87.02%,這與偏差本身主要發(fā)生在生產(chǎn)活動過程中直接相關��。而對于變更控制的監(jiān)管����,盡管MAH是藥品上市后變更的責任主體,但接受調查者認為應由MAH所在地藥品監(jiān)管部門監(jiān)管的僅占35.11%��,這可能是考慮到受托生產(chǎn)企業(yè)是最終實施者�,而MAH所在地藥品監(jiān)管部門難以履行監(jiān)管職能。
4、建議與對策
基于問卷調查的結果��,通過專家專題研討論證�,對偏差處理和變更控制的管理職責,本文最終形成了以下幾點建議��。
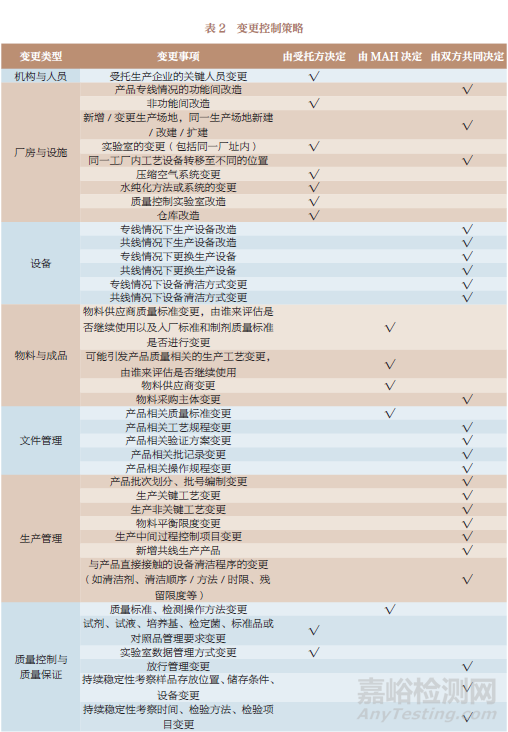
4.1 變更控制策略
對于變更控制的執(zhí)行�,本文按 8個類型的40個事項�,分別給出了建議的控制策略,如受托生產(chǎn)企業(yè)的關鍵人員變更最終應由受托方?jīng)Q定����,而產(chǎn)品專線情況的功能間改造由雙方共同決定,具體建議詳見表2��。

4.2 偏差處理策略
MAH和受托生產(chǎn)企業(yè)雙方對于偏差的處置都較為謹慎�。因此,建議與MAH委托生產(chǎn)的品種相關的偏差發(fā)生時�,均應及時匯報給MAH,由MAH評級��。對于不影響產(chǎn)品安全性�、有效性和質量可控性的微小偏差,由受托方進行記錄��、調查、評估和跟蹤��。在產(chǎn)品放行時����,MAH應當對所有偏差進行審核。對于可能影響產(chǎn)品安全性��、有效性和質量可控性的偏差和檢驗結果偏差��,受托方需要報MAH審核批準�。
4.3 監(jiān)管展望
MAH作為藥品上市許可的責任主體,承擔藥品全生命周期的安全性有效性保證義務����,必須有質量管理能力、風險防控能力�、責任賠償能力。MAH和受托生產(chǎn)企業(yè)應以風險管理為原則�,采取有效措施對偏差和變更進行處理和控制。MAH制度的實施�,使得藥品上市后監(jiān)督管理措施更加有力。對于不在同一區(qū)域的MAH與受托生產(chǎn)企業(yè)�,可采取互聯(lián)網(wǎng)+檢查、聯(lián)合監(jiān)管等模式�。在具體措施上��,通過引入約談��、告誡信�、限制使用��、召回等機制��,根據(jù)雙方質量協(xié)議中的責任劃分�,對MAH和生產(chǎn)企業(yè)追責����,同時追究相關責任人責任。