1995年���,美國食品藥品監(jiān)督管理局(FDA)提出了生物藥劑學(xué)分類系統(tǒng)(BCS)[1]���,即一種科學(xué)框架、將藥物基于其溶解性和滲透性分為 4 類�,目的是為藥品擴(kuò)大生產(chǎn)規(guī)模、上市后的變更(SUPAC)授予生物等效性(BE)豁免�,并以溶出度試驗(yàn)作為判定變更前后是否等效的依據(jù),來替代昂貴耗時(shí)的體內(nèi)研究[2-3]�����。當(dāng)時(shí) BE 豁免僅用于藥品的變更申請(qǐng)。隨后���,BE豁免的范圍被擴(kuò)大至某些口服仿制藥的上市批準(zhǔn)�。FDA于2000年頒布的《基于生物藥劑學(xué)分類的速釋口服固體制劑體內(nèi)生物利用度和生物等效性豁免的指導(dǎo)原則》�,僅活性成分為高溶解性、高滲透性的藥物(BCS I類)且為口服速釋制劑的產(chǎn)品能被豁免���,在 2017年頒布的更新版本[4]中進(jìn)一步將適用范圍擴(kuò)展至活性成分為高溶解性���、低滲透性的藥物(BCS III類)。BE豁免的主要優(yōu)勢在于可以簡化藥品的批準(zhǔn)流程�、減少時(shí)間,從而降低了上市成本���。
經(jīng)檢索中國國家藥品監(jiān)督管理局(NMPA)及其他權(quán)威監(jiān)管機(jī)構(gòu)[如FDA���、歐盟藥品管理局(EMA)、世界衛(wèi)生組織(WHO)]出臺(tái)的有關(guān)人體 BE 豁免的指導(dǎo)原則���,可將 BE 豁免大致分為 2 種類型:第 1 種是基于仿制制劑某一規(guī)格產(chǎn)品的 BE試驗(yàn)顯示與參比制劑生物等效去豁免其他規(guī)格的BE試驗(yàn)���;第2種是基于 BCS 分類豁免某種藥品(所含的原料藥為BCS I類���、BCS III類)的BE試驗(yàn)。
針對(duì)上述 2 種類型的豁免�,中國 NMPA 已出臺(tái)了《以藥動(dòng)學(xué)參數(shù)為終點(diǎn)評(píng)價(jià)指標(biāo)的化學(xué)藥物仿制藥人體BE研究技術(shù)指導(dǎo)原則》[5(] 第1種豁免)、《人體生物等效性試驗(yàn)豁免指導(dǎo)原則》[6(] 第2種豁免)�,F(xiàn)DA 頒布了《工業(yè)指南草案:按簡化新藥申請(qǐng)?zhí)峤坏囊运幋鷦?dòng)力學(xué)為終點(diǎn)的生物等效性研究》[7]、《基于 BCS 分類系統(tǒng)的速釋口服固體劑型的體內(nèi)生物利用度和生物等效性研究》[4]�,其中中國的指導(dǎo)原則主要參考FDA的制定�����;而EMA�����、WHO則將第1種和第 2種類型整合在 1個(gè)指導(dǎo)原則中���,即《生物等效性研究指南(2010 年版)》[8]�����、《WHO 技術(shù)報(bào)告系列第37 號(hào)附件 7:仿制藥實(shí)現(xiàn)其可替代性的注冊(cè)要求指南》[9]�����。
為協(xié)調(diào)統(tǒng)一各成員國監(jiān)管機(jī)構(gòu)的技術(shù)要求�,國際人用藥品注冊(cè)技術(shù)協(xié)調(diào)委員會(huì)(ICH)于 2019 年針對(duì)第 2 種豁免頒布了《M9:基于生物制藥分類系統(tǒng)的生物豁免指導(dǎo)原則》[10],并在問答文件中闡述了該指南允許在仿制藥申請(qǐng)中存在地區(qū)性差異���,以適應(yīng)某些現(xiàn)有法規(guī)不允許仿制藥申請(qǐng)采用基于BCS豁免BE的例外情況�����。中國NMPA及美國FDA均陸續(xù)發(fā)布了實(shí)施該指導(dǎo)原則的通告�,以替代原有的指導(dǎo)原則�����。EMA 在官網(wǎng)發(fā)布了 ICH M9 并表明生效日期為2020年7月3日���,但同時(shí)強(qiáng)調(diào)基于BCS I和 III 類豁免 BE 試驗(yàn)并未在全球內(nèi)達(dá)成共識(shí)�,申報(bào)者需遵循不同地區(qū)的法規(guī)���。日本藥監(jiān)局(PMDA)于2020 年 12 月在官網(wǎng)上公開了 M9 日文稿[11]并表明該指南可適用于部分申請(qǐng)(因參比制劑的處方難以獲得�,暫不適用于仿制藥)�����。綜上,ICH M9 基本替代了各國原有的第2種BE豁免指導(dǎo)原則�。而對(duì)于第 1 種豁免,目前 ICH 正在起草并計(jì)劃于 2024 年完成《M13:速釋口服固體制劑的生物等效性》的制定���,有望對(duì)各國現(xiàn)有的不同指導(dǎo)原則進(jìn)行協(xié)調(diào)統(tǒng)一�����。
上述指導(dǎo)原則適用范圍存在差異之處�����,但均包括了速釋口服固體制劑,WHO�、PMDA 還明確了第1 種豁免亦適用于其他的特殊制劑(如遲釋、緩釋等)及非口服給藥途徑藥物�����,F(xiàn)DA�����、ICH M9提出第2種豁免還適用于新藥開發(fā)。因篇幅有限�����,本文分析的對(duì)象僅針對(duì)申報(bào)量大的速釋口服固體劑型仿制藥�,旨在通過比較國內(nèi)外相關(guān)指導(dǎo)原則對(duì)速釋口服固體劑型的仿制藥藥學(xué)研究的要求,重點(diǎn)關(guān)注存在的差異之處并在最終經(jīng) ICH 協(xié)調(diào)一致的過程中得到思考和啟示�����,了解不同監(jiān)管機(jī)構(gòu)的申報(bào)要求�,以期使得研究者少走彎路,增加仿制藥被豁免體內(nèi)試驗(yàn)的成功率���、進(jìn)而降低開發(fā)成本���,但同時(shí)亦能保證其質(zhì)量和療效與參比制劑一致,真正實(shí)現(xiàn)其臨床可替代性���。
一���、參比制劑的選擇
在仿制藥的注冊(cè)申報(bào)中,均須以參比制劑為標(biāo)桿進(jìn)行相關(guān)的研究�,因此需合理選擇參比制劑�,而關(guān)于參比制劑研究批次的選擇���,不同監(jiān)管機(jī)構(gòu)的要求如下:
(1)日本《仿制藥BE研究的指導(dǎo)原則》[12]規(guī)定�,取3批原研制劑進(jìn)行溶出曲線研究�����,在其規(guī)定的溶出方法下測定���,選取中間那條溶出曲線的批次作為參比制劑樣品�����,若 3 批原研制劑在 15 min 內(nèi)溶出度均能達(dá)到 85%���,則任一批次均可作為參比試劑�����,用于 BE批的參比制劑含量需與標(biāo)示量接近且與仿制制劑含量的差值最好不超過 5%�;
(2)中國要求多批[13(] 通常為 2 批及以上)進(jìn)行藥學(xué)對(duì)比研究,建議BE 批參比制劑含量與仿制制劑含量的差值小于5%[5]�;
(3)EMA 明確用于 BE 試驗(yàn)參比制劑的選擇應(yīng)基于含量(與仿制制劑的含量差值應(yīng)不超過 5%)和溶出數(shù)據(jù)并要求申請(qǐng)人提供資料說明如何基于溶出和含量選擇代表性批次的參比制劑�,建議研究1 批 以 上 的 參 比 制 劑 以 確 定 BE 試 驗(yàn) 用 的 批次[8]�����;
(4)ICH M9征求意見稿中明確了至少 1批�,而在正式稿中則刪除了關(guān)于參比制劑批次的描述[10]。
二�����、對(duì)原料藥的具體要求
第2種BE豁免(基于BCS分類)���,對(duì)原料藥本身的理化性質(zhì)(如結(jié)構(gòu)�����、溶解性)進(jìn)行了限定�����。
其中關(guān)于溶解度的測定各指導(dǎo)原則均明確應(yīng)在pH值1.2~6.8范圍內(nèi)的水溶性介質(zhì)中進(jìn)行�����,EMA和 ICH 還分別說明若藥物的 pKa�、最低溶解度所對(duì)應(yīng)的 pH 值在 1.2~6.8 范圍內(nèi),則需評(píng)價(jià)相應(yīng) pH 值下的溶解度���,而我國及FDA指導(dǎo)原則還明確需考察pH=pKa±1 處的溶解度�。對(duì)于測試藥物的用量�����,我國�����、EMA�����、WHO及ICH指導(dǎo)原則均明確采用最高劑量進(jìn)行試驗(yàn)���,ICH M9 還提出若單次治療的最高劑量不符合高溶解性標(biāo)準(zhǔn)�����,但參比制劑的最高規(guī)格在要求條件下可溶解,則應(yīng)提供額外數(shù)據(jù)以證明基于BCS分類豁免BE的合理性�����。
各指導(dǎo)原則中,ICH M9 最為詳細(xì)地闡述了溶解度的測定方法�����,強(qiáng)調(diào)了研究者容易忽略的試驗(yàn)細(xì)節(jié)問題�,如:
(1)溶解度試驗(yàn)時(shí)間:應(yīng)證明溶解度在預(yù)期吸收時(shí)間范圍內(nèi)(在問答文件中說明了如何確定溶解度測定的持續(xù)時(shí)間)能維持穩(wěn)定。
(2)應(yīng)在添加藥物活性成分后和平衡溶解度研究結(jié)束時(shí)測定每種試驗(yàn)溶液的 pH 值�,以確保溶解度測定是在指定pH值下進(jìn)行,必要時(shí)應(yīng)調(diào)節(jié)pH值�。
(3)將以pH值1.2~6.8內(nèi)測得的最低溶解度對(duì)藥物活性成分進(jìn)行分類。
(4)應(yīng)采用經(jīng)適當(dāng)驗(yàn)證的方法測定溶解度�,使用藥典中適當(dāng)?shù)慕橘|(zhì),在每個(gè)溶解度條件或 pH 值下至少平行測定3個(gè)樣本���。
(5)應(yīng)證明藥物活性成分在溶解介質(zhì)中具有足夠的穩(wěn)定性�,若藥物活性成分在溶解度測定過程中不穩(wěn)定�,即降解>10%,則不能充分確定其溶解度�����,因此無法分類。
在M9問答文件中明確應(yīng)提供試驗(yàn)獲得的溶解度數(shù)據(jù)以確定藥物活性成分的溶解性�。
三、對(duì)制劑處方工藝的要求
3.1 第1種豁免
基于某一規(guī)格生物等效豁免其他規(guī)格 BE 試驗(yàn)���,其他規(guī)格的處方工藝則須滿足一定的前提條件�,下文介紹不同監(jiān)管機(jī)構(gòu)的具體要求�����。
3.1.1 NMPA[5]
若申請(qǐng)仿制制劑的其他規(guī)格 BE豁免�����,則各規(guī)格制劑的處方比例相似�����,是指以下情況:
(1)不同規(guī)格之間所有活性和非活性組分組成比例相似(2022年1月21日發(fā)布的問答文件[14]對(duì)此種情況進(jìn)行了解釋說明)�。
(2)對(duì)于高活性(highpotency)的藥物(活性成分在制劑中所占質(zhì)量比例低):①不同規(guī)格的制劑質(zhì)量一致(差異不超過10%);②各規(guī)格使用相同的非活性組分�;③規(guī)格的變更系通過改變活性組分的用量以及1個(gè)或多個(gè)非活性組分的用量來實(shí)現(xiàn)。
3.1.2 FDA[7]
處方相似性判斷有 3種情況:(1)不同規(guī)格之間所有活性和非活性組分組成比例相似���,如50 mg規(guī)格所有非活性成分為100 mg規(guī)格的1/2�����,或?yàn)?25 mg 規(guī)格的 2 倍���;
(2)符合以下條件:①不同規(guī)格的制劑質(zhì)量幾乎相同(與 BE 規(guī)格的差異不超過10%);②各規(guī)格使用相同的非活性組分���;③規(guī)格的變更系通過改變活性組分的用量以及1個(gè)或多個(gè)非活性組分的用量來實(shí)現(xiàn)���。
(3)不同規(guī)格間活性和非活性成分比例不相似但有充足的證據(jù)可證明處方相似的(FDA將在ANDA評(píng)估期間對(duì)處方相似性進(jìn)行認(rèn)定)。
3.1.3 EMA[8]
(1)相同的制備工藝�;(2)不同規(guī)格處方定性組成相同;(3)不同規(guī)格處方組成比例相似���,如所有規(guī)格的原輔料比例相同(對(duì)于速釋制劑���,包衣成分、膠囊殼�����、色素和香精除外)�。當(dāng)處方等比存在一定誤差時(shí)�����,若滿足以下①和②或①和③時(shí)則仍可判定處方相似���,即可考慮豁免 BE:①活性成分占片芯或膠囊內(nèi)容物質(zhì)量的比不超過 5%;②片芯或膠囊內(nèi)容物中輔料的量與 BE 規(guī)格相同���,僅活性成分的量發(fā)生變化�����;③填充劑的改變量=活性成分的改變量�����,其他輔料的量與BE規(guī)格相同�����。
3.1.4 WHO[9]
不同規(guī)格制備工藝相同且處方相似�,以下 2 種形式均可被認(rèn)為處方相似:
(1)不同規(guī)格之間所有活性和非活性組分組成比例相似(如 50 mg規(guī)格片劑中含有的活性和非活性組分均為 100 mg規(guī)格片劑的 1/2 或者為 25 mg 規(guī)格片劑的 2 倍)���,對(duì)于速釋制劑�,包衣成分、膠囊殼�、色素及香精通常無上述要求;
(2)對(duì)于制劑中原料含量相對(duì)較低的情況(單位劑量最多含 10 mg�����,或質(zhì)量百分比不超過5%)�����,所用規(guī)格間的單位質(zhì)量相似�,若滿足以下條件則可考慮豁免:①片芯或膠囊內(nèi)容物中輔料的量與BE規(guī)格相同���,僅活性成分的量發(fā)生變化���;②填充劑的改變量=活性成分的改變量,其他輔料的量與BE規(guī)格相同�����。
由上可知�����,除 FDA 外,各監(jiān)管機(jī)構(gòu)對(duì)于處方相似性的判斷均分為兩種情況�。第 1 種情況,即不同規(guī)格間原料藥和輔料用量均呈等比增加或減少���;而第 2 種情況�����,各指導(dǎo)原則主要對(duì)原料藥百分比含量的要求略不同�,EMA 明確為占比不超過 5%���,WHO在 EMA 的基礎(chǔ)上增加了另一種情況(即單位劑量含有的活性成分至多10 mg)���,中國于2022年1月21日發(fā)布“對(duì)我國《以藥動(dòng)學(xué)參數(shù)為終點(diǎn)評(píng)價(jià)指標(biāo)的化學(xué)藥物仿制藥人體生物等效性研究技術(shù)指導(dǎo)原則》中關(guān)于多規(guī)格豁免 BE藥學(xué)評(píng)價(jià)標(biāo)準(zhǔn)“處方比例相似性”相關(guān)問題的問答(試行)”[14],在該文件中明確了高活性藥物的標(biāo)準(zhǔn)為占比<5%�。另外,EMA及 WHO 均要求不同規(guī)格與 BE 批次有相同的制備工藝���。
EMA�、WHO 還提出當(dāng)多個(gè)規(guī)格間不符合處方相似的要求時(shí)�����,可采用括號(hào)法,這樣僅進(jìn)行兩端的兩個(gè)規(guī)格(如最高和最低規(guī)格�����,或處方組成差異最大的 2 個(gè)規(guī)格)的體內(nèi) BE 試驗(yàn)即可�����,則剩余規(guī)格組成上的任何差異均可被上述兩個(gè)試驗(yàn)覆蓋���。
3.2 第2種豁免
3.2.1 基本原則
無論基于 BCS I 類還是 III 類豁免 BE試驗(yàn),均對(duì)輔料的選擇有所要求���。ICH M9提出�,理想情況下���,受試制劑的輔料組成應(yīng)模仿參比制劑���。但是,若輔料存在差異�����,應(yīng)評(píng)價(jià)這些差異可能對(duì)體內(nèi)吸收產(chǎn)生的影響。評(píng)價(jià)中應(yīng)考慮藥物活性成分性質(zhì)和輔料作用�。對(duì)于片劑包衣中的較小用量或低于對(duì)特定藥物活性成分產(chǎn)生影響的已知閾值的用量,可相對(duì)較少關(guān)注���。
需注意的是�����,不管是 BCS I 類還是 III 類�,ICHM9均規(guī)定可能影響吸收的輔料應(yīng)種類相同且用量相似���,即在參比制劑輔料用量的±10% 范圍內(nèi)且這些輔料的累計(jì)差異應(yīng)在±10% 范圍內(nèi)�。對(duì)于 BCSIII 類藥物而言�,除了對(duì)影響吸收的輔料有要求外,處方中其他輔料的種類也應(yīng)相同�、用量相似。
WHO 強(qiáng)調(diào)若某藥品中 1 個(gè)規(guī)格基于 BCS 豁免BE 試驗(yàn)���,則其他規(guī)格也必須基于 BCS 分類來評(píng)估是否可以豁免���,而不是基于處方等比;ICH還明確必須每個(gè)規(guī)格均與參比制劑比較來支持基于BCS分類豁免BE。
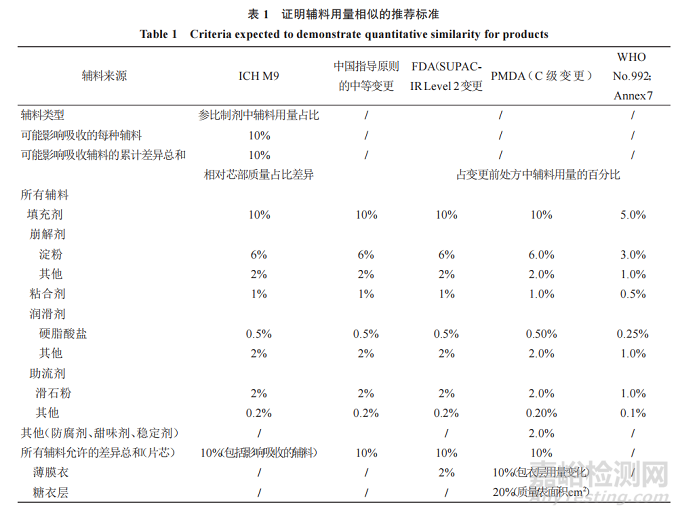
3.2.2 處方相似性判斷
對(duì)基于 BCS分類豁免 BE試驗(yàn)的各指導(dǎo)原則中對(duì)處方相似的判斷標(biāo)準(zhǔn)及各變更指導(dǎo)原則中申請(qǐng)豁免 BE對(duì)處方輔料用量變化的可接受范圍進(jìn)行列表分析(表1)���。FDA基于BCS分類豁免 BE試驗(yàn)的指導(dǎo)原則是由其變更指導(dǎo)原則延伸而來的���,其處方相似性的判斷標(biāo)準(zhǔn)同 SUPACIR Level 2[2-3]的要求,中國現(xiàn)行變更指導(dǎo)原則的中等 變 更[15]�����、ICH M9 及 PMDA[16-17]的 相 關(guān) 規(guī) 定 與FDA 大體是一致的(除對(duì)包衣的要求不同外)�����。而WHO[11]的標(biāo)準(zhǔn)最為嚴(yán)格�����,除未對(duì)包衣膜進(jìn)行限定外�����,其他同 FDA SUPAC-IR Level 1���,亦與我國的微小變更可接受范圍一致。
ICH M9 問答文件[10]解釋:芯部各輔料百分比差異是以相對(duì)仿制制劑與參比制劑各自的芯部進(jìn)行計(jì)算�,若仿制制劑符合這些標(biāo)準(zhǔn)�,但輔料絕對(duì)質(zhì)量存在很大差異(如仿制制劑與參比制劑的芯部質(zhì)量不相似)���,可能須額外論證���。
FDA變更指導(dǎo)原則指出,發(fā)生Level 2變更的藥物屬于高溶高滲(BCS I 類)���、低溶高滲(BCS II 類)或高溶低滲(BCS III類)時(shí)�����,若溶出度等滿足條件可不進(jìn)行 BE 研究�����,否則應(yīng)歸屬于 Level 3���,需進(jìn)行 BE試驗(yàn)(或證實(shí)體內(nèi)外具有相關(guān)性時(shí)可能會(huì)考慮豁免BE)。
中國現(xiàn)行變更指導(dǎo)原則規(guī)定的中等變更與FDA 的 Level 2 變更相比�,除未限定薄膜衣的差異外,其他均一致�����。而中國先前頒布的變更指導(dǎo)原則[18]對(duì)包衣液用量變更進(jìn)行了解釋,即:以原處方單劑量理論質(zhì)量計(jì)算�����,一般允許變更幅度為±2%且包衣液組成不能變化���。PMDA 提出對(duì)于 C 級(jí)變更�����,在藥物的溶解性和治療窗滿足規(guī)定時(shí)�,若溶出等效則可視為生物等效 �,否 則 需 根 據(jù) 仿 制 藥 BE 研 究 指 南[12]進(jìn) 行 BE試驗(yàn)。
總體上來看�,對(duì)于處方相似性的判斷,不同指導(dǎo)原則主要對(duì)包衣膜的要求存在不同�,ICH M9 在FDA 指導(dǎo)原則的基礎(chǔ)上�,刪除了包衣膜用量的控制,可能因?yàn)?BCS I和 III分類藥物制備的速釋制劑溶出均較快�����,受包衣膜影響的可能性較小,且包衣用量的準(zhǔn)確測定亦存在一定困難�����。而日本變更指導(dǎo)原則最大的不同在于其額外明確了含量較少的一些輔料(如防腐劑�����、甜味劑�、穩(wěn)定劑等)的變化量要求,并分別對(duì)薄膜衣和糖衣的組成和包衣質(zhì)量/片芯表面積的變化進(jìn)行了明確規(guī)定(變化水平將片芯和包衣層分開計(jì)算)�,將包衣層中輔料獨(dú)立計(jì)算的原因在于某種情況下包衣層影響藥品的溶出曲線,應(yīng)考慮包衣的厚度而不是包衣層的質(zhì)量�����,因此在指定包衣層可接受變化的標(biāo)準(zhǔn)時(shí)有上述 2 個(gè)指標(biāo)(包衣層組成變化和包衣質(zhì)量/表面積比的變化)�。
另外,對(duì)于輔料不同等級(jí)(或型號(hào))問題���,ICHM9問答文件中說明:若適合�,應(yīng)基于制劑中輔料的功能特性來評(píng)價(jià)輔料等級(jí)的差異�����。對(duì)于一些輔料,等級(jí)改變可能會(huì)影響藥物制劑的溶出(如 HPMC粒度分布���、黏度和取代度的改變�;硬脂酸鹽潤滑劑比表面積的改變)���。對(duì)于輔料相似性的評(píng)價(jià)�,需要具體情況具體分析�,以證明“種類相同”。
關(guān)于處方相似性的計(jì)算方法�����,ICH M9 及日本指導(dǎo)原則中均給出了案例解析�����。
四���、對(duì)溶出曲線的要求
4.1 第1種豁免
各指導(dǎo)原則均明確要求不同規(guī)格制劑的體外溶出曲線相似�,EMA 規(guī)定溶出介質(zhì)為 pH 1.2���、4.5�����、6.8�,WHO 額外增加了質(zhì)量控制(QC)介質(zhì)(被藥典收載的)�����,各介質(zhì)中溶出均需相似(除非經(jīng)論證不滿足漏槽條件)�。當(dāng)不同規(guī)格因在上述介質(zhì)中不滿足漏槽條件導(dǎo)致溶出不相似時(shí),需提供進(jìn)一步研究資料來證實(shí)�����,如通過測試相同劑量的樣品(如2個(gè)5 mg的片劑 vs 1個(gè) 10 mg 的片劑)得出的溶出具備相似性 ���,或經(jīng)試驗(yàn)表明參比制劑具有相同的溶出行為���。
4.2 第2種豁免
各監(jiān)管機(jī)構(gòu)曾實(shí)施的指導(dǎo)原則均對(duì)溶出曲線有具體要求,ICH M9 主要在以下幾個(gè)方面進(jìn)行了協(xié)調(diào)統(tǒng)一�。
(1)溶出介質(zhì)種類 :另增了 1 個(gè)要求 ,即“需在最低溶解度 pH(若與規(guī)定的 3 種緩沖液不同)下進(jìn)行額外研究”�,同時(shí)強(qiáng)調(diào)了不應(yīng)使用有機(jī)溶劑和添加表面活性劑。
(2)溶出介質(zhì)體積:中國及FDA曾實(shí)施的指導(dǎo)原則分別為500 mL或更少�����、500 mL或更少(經(jīng)論證后可選擇 900 mL),ICH M9 統(tǒng)一為“900 mL或更少(建議使用質(zhì)控(QC)檢測所選擇的體積)”
(3)槳法轉(zhuǎn)速:中國�、FDA及WHO允許一定條件下采用75 r·min−1,而ICH M9參照EMA的要求�,僅允許采用50 r·min−1,并在M9問答文件中提到BCS I 類藥物預(yù)計(jì)不會(huì)發(fā)生高變異的情況�����,若發(fā)生變異���、堆積�、黏附�、漂浮等現(xiàn)象 ,建議經(jīng)論證后采用籃 法 100 r·min−1 測 定 �����,而 未 說 明 可 以 提 高 槳 法轉(zhuǎn)速�����。
關(guān)于溶出相似性評(píng)價(jià)�,各國指導(dǎo)原則的要求均相同�����。BCS I 類藥物,仿制制劑與參比制劑需具有快速溶出(30 min 內(nèi) API 的平均溶出百分比均能≥85%)的特點(diǎn)且相似(基于f2比較)���。BCS III類藥物�����,仿制制劑與參比制劑需具有非?��?焖偃艹龅奶攸c(diǎn),即在 15 min 內(nèi) API 的溶出均能達(dá)到標(biāo)示量的 85%以上�����。對(duì)于超過 1 種規(guī)格的制劑�,仿制制劑和參比制劑的每個(gè)規(guī)格均應(yīng)進(jìn)行溶出曲線比較。
ICH M9問答文件中還明確�����,對(duì)于BCS I類藥物預(yù)計(jì)不會(huì)發(fā)生溶出存在高變異的情況�,因此���,不適宜采用替代統(tǒng)計(jì)方法證明相似性(如boost strapping法)。若因堆積效應(yīng)產(chǎn)生高變異�,經(jīng)科學(xué)論證,可考慮替代方法以解決堆積效應(yīng)等問題�。溶出曲線相似性評(píng)價(jià)時(shí),應(yīng)使用仿制制劑和參比制劑的12個(gè)制劑單位所得的溶出數(shù)據(jù)�����,報(bào)告每個(gè)獨(dú)立的制劑單位在每個(gè)特定的時(shí)間點(diǎn)的溶出量(以標(biāo)示量的百分?jǐn)?shù)表示)���,將平均溶出量 �����,溶出范圍(高低值)和變異系數(shù)(相對(duì)標(biāo)準(zhǔn)偏差)列表呈現(xiàn)�����;在進(jìn)行 f2因子計(jì)算時(shí)�,采樣點(diǎn)及相對(duì)標(biāo)準(zhǔn)偏差值需滿足要求�。
五、結(jié)語
在 ICH 文件頒布前,不同監(jiān)管機(jī)構(gòu)有關(guān) BE 豁免的指導(dǎo)原則技術(shù)要求均存在一定的差異���,本文主要對(duì)上述幾個(gè)關(guān)鍵藥學(xué)問題進(jìn)行了探討�����。藥品研發(fā)時(shí)重點(diǎn)關(guān)注不同之處,積極進(jìn)行相關(guān)試驗(yàn)研究���,做出最科學(xué)嚴(yán)謹(jǐn)?shù)倪x擇���。
5.1 參比制劑的選擇
各監(jiān)管機(jī)構(gòu)均明確參比制劑與仿制制劑的含量差值不超過 5%,日本額外強(qiáng)調(diào)了參比制劑的含量需與標(biāo)示量接近�����;而對(duì)參比制劑批次的要求不盡相同�����,但基本上均建議對(duì)多個(gè)批次的參比制劑進(jìn)行研究�。ICH M9終稿中未對(duì)參比制劑的批次進(jìn)行明確,可能是考慮到各國監(jiān)管機(jī)構(gòu)執(zhí)行層面的差異���,未作統(tǒng)一規(guī)定�。若發(fā)現(xiàn)參比制劑的溶出等存在較大的批內(nèi)或批間差異,建議增加批次研究�,對(duì)其質(zhì)量有全面的了解,以選擇合理的批次�。
5.2 處方工藝的要求
5.2.1 處方相似性的判斷
通過對(duì)比不同監(jiān)管機(jī)構(gòu)的指導(dǎo)原則及最終由 ICH 協(xié)調(diào)統(tǒng)一的過程中可以發(fā)現(xiàn),基于 BCS 分類申請(qǐng)豁免 BE 時(shí)輔料用量的可接受標(biāo)準(zhǔn)是由早期 FDA 頒布的變更指導(dǎo)原則發(fā)展而來的�,即基于近 30 年的實(shí)踐積累,而非一蹴而就�����。日本界定藥品的變更等級(jí)時(shí)�����,分別對(duì)包衣膜組成和包衣質(zhì)量與芯部表面積比進(jìn)行了明確規(guī)定�����,并給出了考察包衣對(duì)溶出曲線影響的方法(如采用水楊酸空白片進(jìn)行試驗(yàn))���。研究者可結(jié)合參比制劑的實(shí)際包衣情況(如包衣組成�、增重及包衣質(zhì)量與片芯表面積比)���,考察包衣對(duì)溶出曲線可能存在的影響�����,同時(shí)結(jié)合藥品自身穩(wěn)定性特點(diǎn)(對(duì)光濕等的敏感性)最終確定合理的包衣組成及增重�����,如對(duì)于光不穩(wěn)定的藥物�����,包衣的組成(是否含有遮光劑)及增重對(duì)強(qiáng)光下的穩(wěn)定性影響較為顯著�����;對(duì)于易吸潮的藥 物 ���,包 衣 的 主 要 膜 材 、增 重 及 致 密 性 均 可 能影響片芯對(duì)水分的吸收程度�����。
ICH M9問答文件表明�����,2個(gè)制劑中輔料的絕對(duì)質(zhì)量應(yīng)不存在很大差異,即芯部質(zhì)量需具有相似性�,但未明確具體標(biāo)準(zhǔn),F(xiàn)DA在第1種豁免指導(dǎo)原則中提出片芯質(zhì)量差異在±10% 以內(nèi)認(rèn)為處方相似�����,因此 10% 的標(biāo)準(zhǔn)可作為參考依據(jù)之一�����,但基于 M9中提出的理念“理想情況下�,受試制劑的輔料組成應(yīng)模仿參比制劑。”���,建議研究者盡量選擇與參比制劑相近的芯重�����。亦如 WHO 提到的�����,對(duì)于仿制藥中輔料的選擇�,統(tǒng)一的原則就是,越接近參比制劑���,越容易豁免 BE 試驗(yàn)�。在研發(fā)早期�,可以檢索各監(jiān)管機(jī)構(gòu)參比制劑的審評(píng)報(bào)告、說明書及相關(guān)專利等以獲得全面詳細(xì)的處方信息�,或者運(yùn)用一定的物理化學(xué)方法逆向解析參比制劑的處方,特別是可能影響溶出的輔料���,為了獲得其準(zhǔn)確用量���,建議對(duì)分析方法進(jìn)行必要的驗(yàn)證。
5.2.2 生產(chǎn)工藝
針對(duì)第一種豁免�����,EMA 及 WHO均要求不同規(guī)格與 BE批次有相同的制備工藝�。為避免不同的工藝或關(guān)鍵工藝參數(shù)可能導(dǎo)致的體內(nèi)不等效風(fēng)險(xiǎn)�,建議不同規(guī)格間采用相同的工藝,或參考中國現(xiàn)行的指導(dǎo)原則進(jìn)行相關(guān)研究�����。
5.3 溶出曲線
文中所述的QC方法,WHO解釋為已被藥典收載的方法�,通常為有一定鑒別力的溶出方法,EMA相關(guān)指導(dǎo)原則[19]對(duì)鑒別力的溶出方法定義為“方法可區(qū)分采用不同關(guān)鍵工藝參數(shù)和(或)關(guān)鍵物料屬性(可能對(duì)生物利用度有影響)所生產(chǎn)的不同批次樣品的能力�。理想上所有不等效的批次均可被體外溶出方法監(jiān)測出”。
基于BCS分類豁免BE對(duì)溶出曲線測定條件的要求中須注意到�����,ICH M9 另增加了 1 個(gè) QC 介質(zhì)體積及1個(gè)最低溶解度pH值下的介質(zhì)種類(若與規(guī)定的3種緩沖液不同)���,且不可采用槳法75 r·min−1���。
另外,在中國若新增藥品規(guī)格�,需按補(bǔ)充申請(qǐng)的途徑申報(bào),藥審中心于 2022 年 11 月發(fā)布《〈已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)〉中溶出曲線研究的問答》[21]�����,給出了新增規(guī)格溶出曲線研究的具體建議�����。
本文分析不同監(jiān)管機(jī)構(gòu)先前實(shí)施的法規(guī)與最終協(xié)調(diào)一致的ICH 指導(dǎo)原則存在的差異�,了解指導(dǎo)原則的起草���、頒布及后續(xù)不斷完善的整個(gè)過程,更能深刻的理解其背后的科學(xué)依據(jù)�,對(duì)研發(fā)高質(zhì)量藥品起到事半功倍的作用。同時(shí)提醒申請(qǐng)人注意�����,ICH 僅是在技術(shù)層面進(jìn)行協(xié)調(diào)統(tǒng)一���,其允許在仿制藥申請(qǐng)中存在地區(qū)性差異(如日本因參比制劑處方難以獲得���、暫不適用于仿制藥),EMA明確基于BCS分類的豁免尚未在全球內(nèi)達(dá)成共識(shí)�����,申請(qǐng)人須遵循當(dāng)?shù)胤ㄒ?guī)�。