通過對國內(nèi)外藥品生產(chǎn)設(shè)備清潔驗證的相關(guān)法規(guī)指南全面梳理�����,從質(zhì)量管理體系及驗證工作生命周期管理的角度對清潔驗證進(jìn)行了分析���;明確了藥品生產(chǎn)設(shè)備清潔驗證要點并構(gòu)建了要點結(jié)構(gòu)圖;對國內(nèi)外藥品檢查中發(fā)現(xiàn)的藥品生產(chǎn)設(shè)備清潔驗證存在的缺陷進(jìn)行了統(tǒng)計分析��;對清潔驗證常見問題及分布情況進(jìn)行識別����,為我國藥品生產(chǎn)企業(yè)進(jìn)一步做好藥品生產(chǎn)設(shè)備清潔驗證提供思路與參考,同時也為藥品檢查工作中對清潔驗證針對性檢查提供借鑒�。
最大限度的降低生產(chǎn)過程中的污染和交叉污染是藥品生產(chǎn)質(zhì)量管理最為核心的內(nèi)容之一。在藥品生產(chǎn)過程中�,普遍存在不同藥品共用生產(chǎn)線和生產(chǎn)設(shè)備的情況,如何保證生產(chǎn)設(shè)備的清潔程序能有效去除殘留物及污染物是避免污染與交叉污染的關(guān)鍵點與難點����。而清潔驗證是證明清潔程序能有效清潔設(shè)備并滿足其預(yù)定用途的直接證據(jù)�,也是確保避免污染與交叉污染��,保證最終藥品安全�、有效與質(zhì)量可控的關(guān)鍵內(nèi)容。在藥品生產(chǎn)的歷史上����,不乏由于清潔不徹底導(dǎo)致的藥品質(zhì)量事故[1]。在全球的藥品檢查中也發(fā)現(xiàn)藥品生產(chǎn)企業(yè)關(guān)于生產(chǎn)設(shè)備清潔驗證方面較容易出現(xiàn)缺陷�,其中一些問題還可能影響藥品質(zhì)量,甚至導(dǎo)致藥品安全問題��。如何在質(zhì)量管理體系及驗證生命周期層面做好生產(chǎn)設(shè)備的清潔驗證是保證藥品質(zhì)量的關(guān)鍵內(nèi)容��,有必要進(jìn)行針對性的分析研究��。
1����、藥品生產(chǎn)設(shè)備清潔驗證法規(guī)指南概述及要點結(jié)構(gòu)分析
1.1 藥品生產(chǎn)設(shè)備清潔驗證法規(guī)指南概述
清潔驗證的目的是證明對生產(chǎn)設(shè)備的清潔工作能始終如一的將產(chǎn)品、溶劑及微生物殘留清潔至可接受的限度�,并阻止可能的污染與交叉污染[2]。針對清潔驗證,國內(nèi)外藥品藥品生產(chǎn)質(zhì)量管理規(guī)范(GMP)均進(jìn)行了規(guī)定���,之間雖然存在一些差異�����,但總體原則與要求是一致的��。世界衛(wèi)生組織(WHO)���、美國注射劑協(xié)會(PDA)、原料藥委員會(APIC)���、國際藥品認(rèn)證合作組織(PIC/S)等組織還針對清潔驗證制定了對應(yīng)的指導(dǎo)原則����,如WHO Guidelines on validation-Appendix 3 Cleaning validation(2019) ��、APIC Guidance on aspects of cleaning validation in active pharmaceutical ingredient plants(2016)、PDA Points to Consider for Cleaning Validation(2012)�����、PIC/S Recommendations on Cleaning Validation(2007)。概括起來�,法律法規(guī)及指南最核心的要求是對清潔方法進(jìn)行驗證�,證實其清潔效果����,其中應(yīng)當(dāng)充分評估設(shè)備使用情況���、共線生產(chǎn)情況、清潔方法����、最差條件�、取樣方法與取樣回收率�����、取樣位置、限度標(biāo)準(zhǔn)、殘留物檢驗方法等因素��,并開展必要的持續(xù)確認(rèn)與再驗證����,同時需要關(guān)注標(biāo)準(zhǔn)清潔操作程序文件��、清潔驗證方案、日常監(jiān)測等內(nèi)容。
1.2 基于質(zhì)量管理體系及驗證生命周期的藥品生產(chǎn)設(shè)備清潔驗證要點結(jié)構(gòu)
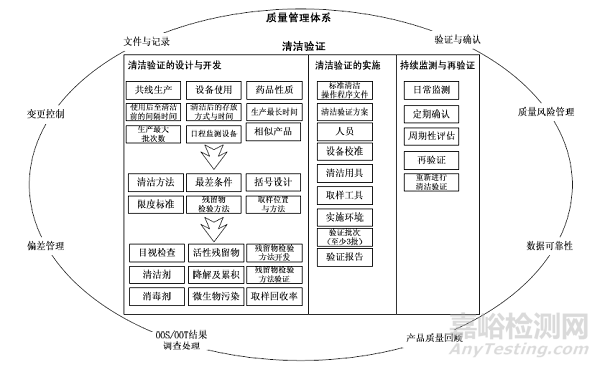
清潔驗證是藥品GMP中明確要求開展的工作�,是確保對生產(chǎn)設(shè)備進(jìn)行有效清潔的重要保證,其必然是藥品生產(chǎn)質(zhì)量管理體系的一項內(nèi)容��。清潔驗證的開展必須遵循質(zhì)量管理體系的各項要求,脫離了質(zhì)量管理體系單獨對清潔驗證進(jìn)行研究容易造成銜接性與體系性方面的問題。從清潔驗證的全過程考慮��,其本身就存在生命周期[3]�����,包括清潔驗證的設(shè)計與開發(fā)�、清潔驗證的實施�����、清潔驗證的持續(xù)監(jiān)測與再驗證3 個階段?��?茖W(xué)、有效的生產(chǎn)設(shè)備清潔驗證必須從質(zhì)量管理體系及生命周期角度對關(guān)鍵要素進(jìn)行管控�����,藥品生產(chǎn)設(shè)備清潔驗證要點結(jié)構(gòu)圖見圖1�����。
圖1 藥品生產(chǎn)設(shè)備清潔驗證要點結(jié)構(gòu)圖
清潔驗證屬于驗證的一種�����,必須遵循驗證的統(tǒng)一要求����,應(yīng)按照驗證主計劃規(guī)定及驗證管理原則實施驗證。在驗證全過程遵循質(zhì)量風(fēng)險管理的要求[4]��,注意文件與記錄的管理,并確保數(shù)據(jù)可靠性����。對實施過程中出現(xiàn)的變更、偏差���、超標(biāo)與超常結(jié)果嚴(yán)格按對應(yīng)程序規(guī)定進(jìn)行管控,并在驗證報告中予以分析描述。在清潔驗證生命周期的3 個階段中,首先是設(shè)計與開發(fā)階段��,該階段涉及的要點最多�����,需要結(jié)合生產(chǎn)線共線生產(chǎn)情況�����、設(shè)備使用情況、原料藥清潔級別[5]���、共線生產(chǎn)藥品性質(zhì)(包括劑型[6]�����、活性物質(zhì)及輔料)開發(fā)清潔方法,制定限度標(biāo)準(zhǔn)���,開發(fā)并驗證活性殘留物檢驗方法���。通過對設(shè)備情況及生產(chǎn)情況分析����,確定取樣位置及取樣方法��,進(jìn)行取樣回收率驗證?��;趯κ褂煤笾燎鍧嵡暗拈g隔時間、生產(chǎn)最長時間�、生產(chǎn)最大批次等內(nèi)容的評估確定最差條件��,根據(jù)相似藥品及設(shè)備的分析明確清潔驗證括號設(shè)計���。其次是驗證實施階段�,該階段在設(shè)計與開發(fā)階段工作的基礎(chǔ)上���,建立對應(yīng)藥品生產(chǎn)設(shè)備標(biāo)準(zhǔn)清潔操作規(guī)程��,制訂清潔驗證方案�����,對從事清潔工作的人員進(jìn)行培訓(xùn)并嚴(yán)格監(jiān)控�。在實施清潔驗證過程中注意對設(shè)備校準(zhǔn)�����、清潔用具�、取樣工具的管理��,確保驗證實施環(huán)境與實際生產(chǎn)及存放環(huán)境一致�,并至少進(jìn)行3 個批次的驗證,最終形成清潔驗證報告�。最后是持續(xù)監(jiān)測與再驗證����,在完成清潔驗證的實施,確認(rèn)清潔方法有效后�,仍需要進(jìn)行持續(xù)的監(jiān)測����,確保清潔方法的持續(xù)有效[7]����,制定日常清潔監(jiān)測標(biāo)準(zhǔn)(如目視檢查���、pH 值�����、電導(dǎo)率及總有機碳[8]監(jiān)測等)����,通過定期確認(rèn)及周期性的評估確保其有效運行,必要時進(jìn)行再驗證����。特殊情況下(如引入新的產(chǎn)品���、清潔方法變更等),可能需要重新進(jìn)行清潔驗證����。
1.3 藥品生產(chǎn)設(shè)備清潔驗證關(guān)鍵要點分析
在清潔驗證要點結(jié)構(gòu)框架中����,存在一些較常出現(xiàn)問題及相對較復(fù)雜的要點值得重點關(guān)注���,主要包括:
(1)限度標(biāo)準(zhǔn):包括目視檢查、微生物負(fù)荷��、活性殘留物�����、清潔劑及消毒劑殘留�、降解物殘留�����、殘留物累積���、其他可能的毒性成分殘留(如沙坦類藥品中的亞硝胺雜質(zhì)�、回收溶劑可能帶入的雜質(zhì)等)的限度標(biāo)準(zhǔn)規(guī)定��,其中活性殘留物的選擇是最為核心的內(nèi)容�����?;钚詺埩粑锟梢葬槍γ總€產(chǎn)品建立,也可從相似品種中選擇最差條件的產(chǎn)品作為目標(biāo)殘留物�����,也可基于溶解性��、毒性���、活性、清潔難度及可檢測情況基于風(fēng)險選擇一種或多種目標(biāo)殘留物�,并通過對日常治療劑量的0.1%��、1×10−5 及基于可接受日暴露量(ADE)及允許日暴露量(PDE)等安全性指標(biāo)計算的限度標(biāo)準(zhǔn)三者中選擇最低的值作為活性殘留物限度標(biāo)準(zhǔn)[9-10]��,其中對于中藥生產(chǎn)應(yīng)考慮其特殊性[11]���。
(2)取樣:包括取樣方法�,取樣位置(基于設(shè)備設(shè)計與復(fù)雜程度)[12]��、取樣工具、取樣人員�、取樣操作[13]、取樣后樣品處理����、回收率確認(rèn)、取樣設(shè)備表面材質(zhì)等需要關(guān)注的內(nèi)容�。取樣方法應(yīng)包括直接取樣(棉簽擦拭)和淋洗取樣�,并針對不同的取樣位置選擇適宜的取樣方法�,在選擇取樣位置時需要注意包括最難清洗位置[14]����,并考慮對連接管道等輔助設(shè)備的取樣�����。開展取樣方法�、取樣設(shè)備表面材質(zhì)及取樣人員回收率確認(rèn)(至少大于50%)����,并注意對取樣棉簽材質(zhì)及供應(yīng)商的管控��、取樣后樣品的處理�、現(xiàn)場取樣面積模具配備、回收率驗證模擬過程與實際情況的一致性等內(nèi)容���。
(3)清潔方法:包括同一產(chǎn)品更換批次的清潔方法(小清)、更換不同產(chǎn)品時的清潔方法(大清)[15]����、針對各品種的清潔方法、針對不同設(shè)備的清潔方法����、清潔程序建立依據(jù)、清潔方式(手動清潔����、半自動清潔及自動清潔)、具體的清潔程序(如清潔劑����、濃度����、溫度��、體積���、清潔次數(shù)����、清潔時間�、設(shè)備運行參數(shù)設(shè)置���、人員操作����、清潔步驟)等�。
(4)最差條件:應(yīng)充分評估生產(chǎn)最長時間、最大批次數(shù)��、使用后至清潔開始的最長時間間隔����、清潔后的存放方式與時間�、降解與累積的風(fēng)險等。
(5)檢驗方法:活性殘留物檢測方法必須經(jīng)過驗證����,檢測限、定量限�、線性范圍及專屬性等應(yīng)適用于活性殘留限度標(biāo)準(zhǔn)�,如亞硝胺殘留在不同產(chǎn)品中的殘留情況檢測,不能直接套用美國食品藥品監(jiān)督管理局(FDA)發(fā)布的用于檢測纈沙坦中是否存在雜質(zhì)N-亞硝基二甲胺雜質(zhì)的氣相色譜–質(zhì)譜(GC/MS)頂空法[16]�。
2�����、我國藥品檢查中藥品生產(chǎn)設(shè)備清潔驗證缺陷的識別和分析
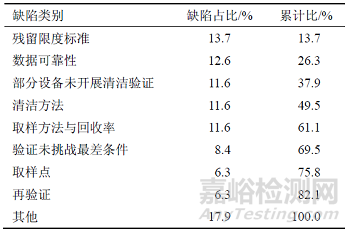
對于存在多產(chǎn)品共線生產(chǎn)的企業(yè)�,現(xiàn)場檢查的重點必然包括清潔驗證��。通過對國家核查中心2017—2018 年跟蹤檢查中發(fā)現(xiàn)的缺陷進(jìn)行統(tǒng)計分析,發(fā)現(xiàn)涉及清潔驗證的缺陷共78 條��,主要問題及分布情況見表1����。
表1 2017—2018 年跟蹤檢查清潔驗證缺陷分布情況
清潔驗證的各類缺陷中��,居于首位的是殘留限度標(biāo)準(zhǔn)的問題�,包括殘留檢驗項目不合理(如僅對清潔劑殘留進(jìn)行檢測�����、僅進(jìn)行目視殘留檢測����、設(shè)備堿液浸泡清洗后不測試pH值等)���,具體限度數(shù)值(如紫外吸光度小于特定數(shù)值等)制定依據(jù)不充分���,未按驗證方案規(guī)定項目進(jìn)行全項檢測等。其次是數(shù)據(jù)可靠性方面的缺陷��,主要包括清潔驗證報告及所附清潔記錄內(nèi)容及信息存在缺失或錯誤(如設(shè)備清潔記錄未記錄清洗方法與過程,遺漏部分設(shè)備清潔驗證數(shù)據(jù)��、設(shè)備型號信息錯誤等)���,清潔驗證樣品檢測數(shù)據(jù)處理不規(guī)范(手動積分����、禁止積分參數(shù)設(shè)置等)�����。其三是部分設(shè)備未開展清潔驗證���,除部分設(shè)備沒有進(jìn)行清潔驗證外��,還存在未對非專用濾芯、鏈接管道等進(jìn)行清潔驗證的情況�。清潔方法的主要問題包括清潔方法未考慮目標(biāo)物質(zhì)的溶解性����,清潔程序規(guī)定(如淋洗用溶媒量)不具體��,缺少對手工清潔操作中人員、時間等影響因素的評估����。取樣方法與回收率方面的問題主要包括取樣方法僅使用淋洗法取樣(未使用擦拭法)��,取樣規(guī)定可操作性差(如對取樣面積��、取樣方式���、圖示等描述不明確),未對取樣人員進(jìn)行取樣回收率驗證�����,取樣回收率驗證溶液濃度不在可接受范圍內(nèi)等���。驗證未挑戰(zhàn)最差條件的缺陷中,主要是清潔驗證中未對更換品種���、生產(chǎn)結(jié)束至清潔開始的最長時間、最長清潔周期進(jìn)行驗證��。取樣點的問題集中在最難清潔點選擇及已選擇取樣點的代表性方面����。再驗證的方面主要是文件中規(guī)定的再驗證周期缺少合理依據(jù)及未按規(guī)定進(jìn)行定期再驗證的問題���。此外,對于清潔驗證中發(fā)生的偏差�����,對清潔驗證涉及的共線風(fēng)險評估報告����、分析方法驗證及驗證批數(shù)等方面也發(fā)現(xiàn)一些問題的存在���。
除在對藥品生產(chǎn)企業(yè)的GMP 跟蹤檢查外����,在藥品注冊生產(chǎn)現(xiàn)場核查中����,新申報產(chǎn)品引入已有生產(chǎn)線的清潔驗證情況也是核查重點���,其中包括新產(chǎn)品對原有產(chǎn)品的影響�,也包括原有產(chǎn)品對新引入產(chǎn)品的影響����。在近幾年的注冊生產(chǎn)現(xiàn)場核查中�,曾出現(xiàn)由于企業(yè)共線生產(chǎn)細(xì)胞毒性藥品的清潔驗證不完善�,存在污染與交叉污染的風(fēng)險等問題,導(dǎo)致產(chǎn)品未能獲批的情況����。
3���、境外藥品檢查中藥品生產(chǎn)設(shè)備清潔驗證缺陷的識別和分析
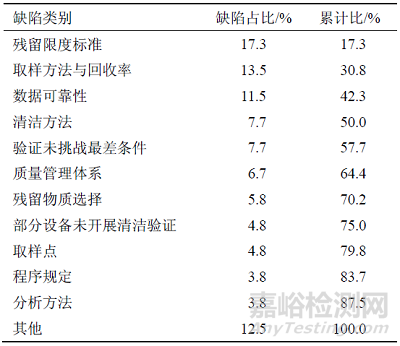
近年來�,基于監(jiān)管機構(gòu)間的合作協(xié)議�,我國藥品監(jiān)督管理部門也會應(yīng)境外藥品檢查及監(jiān)管機構(gòu)(如世界衛(wèi)生組織�、美國食品藥品監(jiān)督管理局等)的邀請,觀察其對我國境內(nèi)部分藥品生產(chǎn)企業(yè)的檢查�。2015—2018 年觀察檢查中記錄的關(guān)于清潔驗證的缺陷共72 條����,見表2。
表2 2015—2018 年境外觀察檢查中清潔驗證缺陷分布情況
與我國藥品檢查相比�,境外藥品檢查觀察在清潔驗證方面的缺陷類型基本一致,除各類缺陷的占比存在一些差異外,在缺陷內(nèi)容方面也存在一些區(qū)別�����,主要包括:
(1)在殘留限度標(biāo)準(zhǔn)方面�,對微生物檢測����、1×10−5 標(biāo)準(zhǔn)的合理性、依據(jù)ADE 和PDE計算殘留限度的要求���、TOC 標(biāo)準(zhǔn)與實際殘留量的關(guān)系、累積與降解情況方面的發(fā)現(xiàn)的問題相對較多��;
(2)在取樣方法與回收率方面����,對回收率驗證涉及的材質(zhì)(如密封膠圈�����、管道�、聚丙烯��、玻璃等)、擦拭測定中樣品干燥條件與實際工藝中干燥條件的一致性�����、擦拭棉簽浸潤及浸泡溶劑����、取樣工具與模具的管理也更為關(guān)注���;
(3)數(shù)據(jù)可靠性方面����,關(guān)注限度標(biāo)準(zhǔn)計算過程的記錄及驗證方案報告的規(guī)范管理�;
(4)清潔方法方面,還發(fā)現(xiàn)實際清潔操作與驗證的清潔方法不一致��,存在額外清潔性操作(如清潔后使用有機溶媒進(jìn)行表面潤洗)未納入驗證���,不同程序規(guī)定的清潔方法不一致,清潔用具管理不規(guī)范的問題�;
(5)驗證未挑戰(zhàn)最差條件方面,對最大連續(xù)生產(chǎn)批次數(shù)進(jìn)行驗證及評估的問題;
(6)質(zhì)量管理體系方面�,主要是清潔驗證過程發(fā)生的偏差處理、變更控制�、超標(biāo)結(jié)果調(diào)查中存在不足;
(7)殘留物質(zhì)選擇方面����,包括活性物質(zhì)矩陣表缺少對新引入物質(zhì)的評估、未考慮溶劑殘留���、選擇的活性物質(zhì)不能代表其他產(chǎn)品等情況;
(8)部分設(shè)備未開展清潔驗證方面����,同時也關(guān)注輔助生產(chǎn)設(shè)備(如灌裝計量槍、濾袋�、中轉(zhuǎn)桶)的清潔驗證����。此外,對于依據(jù)歐盟清潔驗證規(guī)定的要求��、殘留分析方法驗證�、清潔驗證中的設(shè)備分類等方面也發(fā)現(xiàn)了一些問題。
通過對2015—2019 年FDA 針對我國境內(nèi)藥品生產(chǎn)企業(yè)發(fā)出的77 封警告信分析發(fā)現(xiàn)�,涉及清潔驗證缺陷的問題主要包括:偽造清潔驗證報告的數(shù)據(jù)可靠性問題、清潔驗證不充分�、部分設(shè)備未開展清潔驗證、未對清潔驗證分析檢測中出現(xiàn)的異常峰進(jìn)行調(diào)查���。
4、結(jié)語
科學(xué)�����、有效的藥品生產(chǎn)設(shè)備清潔驗證是避免產(chǎn)生藥品污染與交叉污染的重要措施����,特別是在多產(chǎn)品共線生產(chǎn)的情況下�,直接關(guān)系到藥品質(zhì)量與安全,做好藥品生產(chǎn)設(shè)備的清潔驗證對藥品生產(chǎn)企業(yè)至關(guān)重要���。在實際工作中,藥品生產(chǎn)企業(yè)可以參照本文提出的藥品生產(chǎn)設(shè)備清潔驗證要點結(jié)構(gòu)圖對清潔驗證開展情況進(jìn)行分析�,借鑒國內(nèi)外藥品檢查中常見的缺陷進(jìn)行對照,依據(jù)國內(nèi)外相關(guān)法規(guī)指南�,結(jié)合實際情況不斷完善���、改進(jìn)清潔驗證工作���。在藥品檢查中����,藥品檢查員可以參考清潔驗證要點結(jié)構(gòu)圖及常見缺陷情況����,基于風(fēng)險開展針對性的檢查,進(jìn)一步提升現(xiàn)場檢查質(zhì)量和效率�。
參考文獻(xiàn)
[1] 薛 峰. 關(guān)于藥品GMP 檢查中清潔驗證常見問題的矯正 [J]. 藥學(xué)與臨床研究, 2020, 28(1): 74-77.
[2] WHO. Good manufacturing practices: guidelines on validation. Appendix 3. Cleaning validation [EB/OL]. [2020-05-03]. https://extranet.who.int/prequal/content/inspections-0.
[3] 翟鐵偉. 藥品生產(chǎn)中清潔驗證的生命周期探討 [J]. 中國醫(yī)藥工業(yè)雜志, 2019, 50(11): 1341-1347.
[4] 姜 彬. 質(zhì)量風(fēng)險管理在非無菌原料藥清潔驗證評估中的應(yīng)用 [J]. 中國藥師, 2017, 20(12): 2281-2285.
[5] APIC. Guidance on aspects of cleaning validation in active pharmaceutical ingredient plants [EB/OL].(2016-09-12) [2020-05-03].
[6] 郝瑩華, 梁 毅. 原料藥生產(chǎn)設(shè)備清潔驗證的范圍和程度評估 [J]. 中國醫(yī)藥工業(yè)雜志, 2018, 49(4): 522-527.
[7] 熊 浪, 梁 毅. 基于生命周期理論的共線生產(chǎn)清潔驗證關(guān)鍵點研究 [J]. 制藥裝備, 2016, 12(12): 8-12.
[8] 莊目德. 清潔驗證TOC(總有機碳)取樣回收率研究[J]. 海峽藥學(xué), 2017, 29(7): 73-75.
[9] EMA. Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities [EB/OL].(2014-12-20) [2020-05-05].
[10] EMA. Questions and answers on implementation of riskbased prevention of cross-contamination in production and guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities [EB/OL]. (2018-04-19) [2020-05-05].
[11] 王守斌, 肖傳學(xué), 黃雪紅, 等. 多品種共線的中藥制劑清潔驗證 [J]. 中草藥, 2016, 47(10): 1815-1819.
[12] 柴振平, 高 鵬, 白亞靈, 等. 化學(xué)藥片劑生產(chǎn)設(shè)備清潔方法的驗證 [J]. 中國藥房, 2015, 26(34): 4755-4758.
[13] 馬肖夢, 黃麗敏, 許漢林. 關(guān)于設(shè)備清潔殘留物限度驗證的探討 [J]. 湖北中醫(yī)雜志, 2016, 38(8): 69-72.
[14] 高 歌, 張會云, 尤春玲, 等. 制藥企業(yè)共線生產(chǎn)產(chǎn)品清潔驗證 [J]. 質(zhì)量探索, 2016, 138(4): 46-47.
[15] 王守斌, 聶 杰, 陳如柳, 等. 淺析藥品生產(chǎn)設(shè)備的清潔驗證 [J]. 天津藥學(xué), 2014, 26(5): 72-76.
[16] FDA. FDA-published testing method to provide an option for regulators and industry to detect NDMA impurities[EB/OL]. (2019-10-17) [2020-05-04].