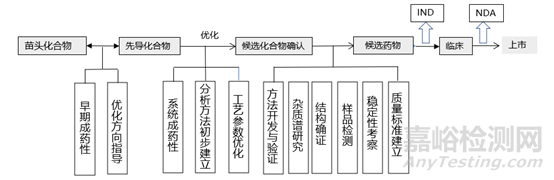
創(chuàng)新藥研發(fā)需要經(jīng)過多個(gè)研究階段,其中申報(bào)IND是第一個(gè)重要節(jié)點(diǎn)���,也是創(chuàng)新藥邁向成功的第一步���,IND階段的研究是后續(xù)臨床研究的重要基礎(chǔ)�����。
IND階段的研究工作包括藥學(xué)研究(CMC)、臨床前的毒理、藥效�、藥代、藥動(dòng)學(xué)等研究���。其中藥學(xué)研究是藥物研發(fā)的重要組成部分���,對(duì)非臨床和后續(xù)臨床研究試驗(yàn)提供技術(shù)和物質(zhì)支持�����,它由三個(gè)部分組成:原料藥(C)、制劑(M)�����、質(zhì)量(C)。其中質(zhì)量是銜接原料藥和制劑的橋梁�,且貫穿新藥研發(fā)的全生命周期�����,是安全性���、有效性的重要保障�。
那么在IND申報(bào)階段需要開展哪些質(zhì)量相關(guān)的工作呢���?其實(shí)通過解讀指導(dǎo)原則不難總結(jié)出需要開展哪些工作�����,但是具體怎樣開展���?需要注意哪些細(xì)節(jié)指導(dǎo)原則并沒有給給出答案�,本文的目的是以指導(dǎo)原則為前提���,結(jié)合自己的經(jīng)驗(yàn)談一談在創(chuàng)新藥IND申報(bào)階段質(zhì)量研究的工作內(nèi)容及相關(guān)實(shí)施策略�����。
創(chuàng)新藥IND申報(bào)階段質(zhì)量研究的內(nèi)容
1.1 原料藥質(zhì)量研究
原料藥和制劑是質(zhì)量研究的兩個(gè)媒介基礎(chǔ)�,原料藥質(zhì)量研究是藥學(xué)研究工作開展以及藥效和安全性評(píng)價(jià)工作開展的重要物質(zhì)基礎(chǔ)。在創(chuàng)新藥IND申報(bào)階段�,原料藥質(zhì)量研究的具體工作有以下幾個(gè)部分。
1.1.1類藥性考察
新藥研發(fā)過程中,候選化合物的確認(rèn)是一項(xiàng)具有重大意義的里程碑�����。不難想象�,一個(gè)化合物從苗頭化合物�,到先導(dǎo)化合物���,在再到確認(rèn)為候選藥物并申報(bào)I其臨床研究需要經(jīng)過一個(gè)迂回反復(fù)的過程和遞進(jìn)式的篩選���,在這個(gè)過程中各個(gè)專業(yè)研究人員各顯其能���,從各自專業(yè)的角度從眾多化合物中篩選出最可能成為藥物的化合物�����,也就是開展類藥性評(píng)價(jià)。在研發(fā)早期�����,類藥性是篩選化合物的重要指標(biāo),是將理化性質(zhì)與體內(nèi)行為聯(lián)系起來的重要手段。
(1)溶解性:考察化合物在不同pH緩沖液中的溶解度���,以對(duì)化合物在體內(nèi)的吸收情況有初步預(yù)測(cè)�。pH設(shè)置通常模擬并覆蓋藥物在體內(nèi)吸收所涉及的各個(gè)部位的pH���,如pH1.2�����、4.5、6.8�、7.4是常常需要考察的pH。
(2)親脂性:化合物的脂溶性直接影響化合物的透膜性�����、溶解性���、毒性�����、分布容積和蛋白結(jié)合率���,是類藥性的重要指標(biāo)�。在開發(fā)早期考察化合物親脂性���,尤其是logD(6.5&7.4)可以對(duì)化合物的ADME進(jìn)行初步預(yù)測(cè)。
(3)解離常數(shù):通過化合物解離常數(shù)可以判斷化合物在不同的吸收部位是分子狀態(tài)還是離子狀態(tài)���,從而可以粗略預(yù)判化合物在不同的吸收部位的吸收情況�����。同時(shí)也可以根據(jù)化合物的解離常數(shù)對(duì)排泄器官pH進(jìn)行調(diào)節(jié)從而調(diào)節(jié)化合物的排泄�����。
(4)穩(wěn)定性:對(duì)化合物的固有穩(wěn)定性進(jìn)行初步考察�����,包括高溫���、高濕�����、光照條件下的穩(wěn)定性���、加速穩(wěn)定性和長(zhǎng)期穩(wěn)定性���,對(duì)于穩(wěn)定性明顯較差的化合物可以根據(jù)穩(wěn)定性的程度設(shè)計(jì)不同程度的考察條件。穩(wěn)定性的考察結(jié)果不僅對(duì)后續(xù)的儲(chǔ)存運(yùn)輸有一定的指導(dǎo)意義���,從穩(wěn)定性的角度也可以對(duì)一系列的待篩選化合物進(jìn)行評(píng)分�,排除不適宜繼續(xù)開發(fā)的化合物�。例如化合物A和B藥效�����、安全性���、親脂性、溶解性等指標(biāo)均相近���,但A的穩(wěn)定性明顯差于B,那么經(jīng)過穩(wěn)定性考察可以將A排除�����。
1.1.2分析方法初步開發(fā)
研究早期分析方法開發(fā)注重通用性���,分析方法可以適當(dāng)粗糙�,能夠?qū)Σ煌瑯悠愤M(jìn)行評(píng)比打分即可�����。該方法可用于早期藥理試驗(yàn)用樣品檢測(cè)和后續(xù)的工藝路線選擇與工藝優(yōu)化。
1.1.3藥理試驗(yàn)樣品檢測(cè)
在候選化合物基本確定后���,會(huì)開展化合物的毒理和藥效試驗(yàn),采用初步建立的分析方法考察藥理試驗(yàn)用樣品是否滿足藥理試驗(yàn)要求�。
1.1.4工藝路線選擇與優(yōu)化
對(duì)不同路線和工藝下的產(chǎn)品進(jìn)行檢測(cè),從產(chǎn)品雜質(zhì)譜和雜質(zhì)含量的角度對(duì)路線進(jìn)行選擇���,對(duì)工藝參數(shù)進(jìn)行優(yōu)化�����。
1.1.5質(zhì)量控制
1.1.5.1起始物料質(zhì)量控制
“原料藥的起始物料”是指其關(guān)鍵結(jié)構(gòu)將進(jìn)入原料藥中的一種原料�、中間體或用來生產(chǎn)該原料藥的一種活性原料成分,原則上市售的���、按合同或商業(yè)協(xié)議從一個(gè)或多個(gè)供應(yīng)商處購得的或者企業(yè)自制的都可以作為起始物料。多數(shù)情況下���,起始物料都是從供應(yīng)商處購買的�����。在起始原料質(zhì)量控制方面的工作有以下幾項(xiàng):
(1)輔助確定生產(chǎn)商:對(duì)不同廠家起始物料和相應(yīng)的終產(chǎn)品進(jìn)行檢測(cè)���,考察不同廠家起始物料對(duì)終產(chǎn)品質(zhì)量的影響�����,輔助確定生產(chǎn)商���。
(2)控制策略制定:大多數(shù)創(chuàng)新藥的反應(yīng)步驟不多(通常為3步反應(yīng))�,因此起始物料的質(zhì)量往往會(huì)直接影響終產(chǎn)品質(zhì)量以及工藝的穩(wěn)定性。起始物料的質(zhì)量控制首先考察COA報(bào)告中的質(zhì)控項(xiàng)目和限度的設(shè)置是否能滿足穩(wěn)定產(chǎn)生符合要求的終產(chǎn)品�����,是否需要添加對(duì)終產(chǎn)品質(zhì)量影響較大的重要控制項(xiàng)目���,而對(duì)于那些對(duì)終產(chǎn)品質(zhì)量無顯著影響的項(xiàng)目可以酌情刪除�。總之���,直接采用COA還是建立內(nèi)控標(biāo)準(zhǔn),應(yīng)視起始物料的各控制項(xiàng)目對(duì)后續(xù)反應(yīng)和終產(chǎn)品質(zhì)量的影響而決定�����。
(3)雜質(zhì)的控制:對(duì)于DNA反應(yīng)性雜質(zhì)�、對(duì)后續(xù)反應(yīng)或終產(chǎn)品質(zhì)量有影響的雜質(zhì)、殘留溶劑一�、二類溶劑和一些毒性較大的無機(jī)雜質(zhì)(如重金屬)需要根據(jù)工藝路線和雜質(zhì)在反應(yīng)中的清除情況來確定控制策略���。如果具有手性中心,要把異構(gòu)體雜質(zhì)作為關(guān)鍵雜質(zhì)進(jìn)行控制�����。對(duì)于普通有機(jī)雜質(zhì)可以依據(jù)COA的限度來加以控制�。三類殘留溶劑和普通無機(jī)雜質(zhì)通常在終產(chǎn)品中進(jìn)行控制�����,在起始物料中可以不設(shè)置控制項(xiàng)。
(4)結(jié)構(gòu)確證:對(duì)起始物料和關(guān)鍵雜質(zhì)進(jìn)行結(jié)構(gòu)確證(質(zhì)譜���、核磁)
(5)分析方法優(yōu)化和驗(yàn)證:首先考察起始物料廠家提供的COA報(bào)告中的方法是否適用�����,能否滿足我們預(yù)設(shè)的靈敏度和專屬性���,如果滿足,可以直接采用COA報(bào)告中的分析方法���,否則,需要對(duì)方法進(jìn)行優(yōu)化���。以COA提供的方法為基礎(chǔ),適當(dāng)對(duì)色譜柱�����、流動(dòng)相pH、緩沖鹽種類�����、緩沖鹽濃度或酸濃度�����、流動(dòng)相梯度等條件進(jìn)行篩選和優(yōu)化���。對(duì)優(yōu)化所得的分析方法進(jìn)行驗(yàn)證�����,在IND申報(bào)階段,進(jìn)行專屬性、靈敏度和溶液穩(wěn)定性驗(yàn)證即可�����。
(6)樣品檢測(cè)和數(shù)據(jù)積累�����。
1.1.5.2中間體
中間體是原料藥工藝合成過程中產(chǎn)生的一種物料,經(jīng)過進(jìn)一步分子變化或精制可以成為原料藥�。中間體尤其是關(guān)鍵中間體質(zhì)量對(duì)成品質(zhì)量有顯著影響�。在中間體質(zhì)量控制過程中需要完成的工作包括以下幾項(xiàng):
(1)控制策略制定:中間體中可能存在起始物料、副產(chǎn)物或降解產(chǎn)物�����,對(duì)于那些含量較高或可對(duì)后續(xù)反應(yīng)和終產(chǎn)品有明顯影響的雜質(zhì)應(yīng)進(jìn)行詳細(xì)研究和控制。最后���,依據(jù)合成工藝,結(jié)合對(duì)工藝過程中的關(guān)鍵控制點(diǎn)的認(rèn)識(shí)�,制定中間體的內(nèi)控標(biāo)準(zhǔn)�。
(2)結(jié)構(gòu)確證:對(duì)中間體和關(guān)鍵雜質(zhì)進(jìn)行結(jié)構(gòu)確證(質(zhì)譜�、核磁)�。
(3)分析方法開發(fā):依據(jù)中間體質(zhì)量控制的需求�,開發(fā)相應(yīng)的分析方法���,通常涉及有關(guān)物質(zhì)方法的開發(fā)�。
(4)分析方法驗(yàn)證:中間體分析方法驗(yàn)證僅進(jìn)行部分關(guān)鍵驗(yàn)證即可�,包括專屬性�、靈敏度和溶液穩(wěn)定性。
(5)樣品檢測(cè)和數(shù)據(jù)積累�����。
(6)質(zhì)量標(biāo)準(zhǔn):依據(jù)合成工藝和樣品檢測(cè)數(shù)據(jù)統(tǒng)計(jì)�����,建立中間體的內(nèi)控標(biāo)準(zhǔn)�,控制限度可以適當(dāng)放寬�����。
1.1.5.3成品
(1)控制策略制定
原料藥控制項(xiàng)目可分為一般控制項(xiàng)目和個(gè)性化控制項(xiàng)目:一般控制項(xiàng)目如性狀、鑒別���、含量���、有關(guān)物質(zhì)���,個(gè)性化控制項(xiàng)目根據(jù)產(chǎn)品擬定的給藥方式和自身特點(diǎn)來設(shè)置���,如口服制劑的晶型和粒度,注射液的細(xì)菌內(nèi)毒素和不溶性微粒等�����。依據(jù)ICH 指導(dǎo)原則原料藥中雜質(zhì)可分為殘留溶劑�、無機(jī)雜質(zhì)和有機(jī)雜質(zhì)。其中有機(jī)雜質(zhì)就是所熟知的有關(guān)物質(zhì)���,是影響產(chǎn)品質(zhì)量的重要因素���。有關(guān)物質(zhì)來源包括起始物料�、中間體及及其關(guān)鍵雜質(zhì)的殘留、降解產(chǎn)物�����、副產(chǎn)物�、異構(gòu)體轉(zhuǎn)化等�。對(duì)于創(chuàng)新藥中的有關(guān)物質(zhì)�,一般雜質(zhì)限度可依據(jù)ICH Q3A制定�,特定雜質(zhì)限度需根據(jù)雜質(zhì)安全性、工藝可行性和產(chǎn)品穩(wěn)定性綜合制定���。對(duì)于DNA反應(yīng)性雜質(zhì)根據(jù)ICH M7指導(dǎo)原則進(jìn)行控制;無機(jī)雜質(zhì)中需重點(diǎn)關(guān)注的是金屬類雜質(zhì)���,其控制限度參考ICH Q3D制定;殘留溶劑控制限度參考ICH Q3C制定�����。對(duì)于雜質(zhì)的控制方式可以有幾種不同的方式:一是終點(diǎn)控制,二是中控檢測(cè)+終點(diǎn)控制�,三是基于對(duì)工藝參數(shù)及其對(duì)殘留雜質(zhì)水平(包括去向和清除知識(shí))的影響的了解���,確信原料藥中的相應(yīng)的雜質(zhì)水平將會(huì)低于可接受限度���,并有多批數(shù)據(jù)作為支撐�,對(duì)相應(yīng)雜質(zhì)不做控制�����。
(2)結(jié)構(gòu)確證和對(duì)照品標(biāo)化
對(duì)原料藥進(jìn)行全面結(jié)構(gòu)分析和確證包括UV�、IR���、MS、NMR(全套)���、XRD�����、MXRD�、EA、DSC等���。當(dāng)存在異構(gòu)體雜質(zhì)時(shí)���,應(yīng)對(duì)異構(gòu)體雜質(zhì)進(jìn)行部分結(jié)構(gòu)確證,包括MS�����、NMR(全套)���、MXRD�����,其他雜質(zhì)進(jìn)行MS和NMR結(jié)構(gòu)確證�����。同時(shí)對(duì)原料藥及雜質(zhì)對(duì)照品進(jìn)行標(biāo)化���。對(duì)于多肽化合物還需提供氨基酸序列���、氨基酸組成分析�����、高級(jí)結(jié)構(gòu)確定等信息���。
(3)理化性質(zhì)研究
理化性質(zhì)研究包括兩部分�,一部分是普通理化性質(zhì)�����,包括性狀�����、熔點(diǎn)�����、比旋度�,引濕性等���,一部分是關(guān)鍵理化性質(zhì)�����,通常與制劑性能相關(guān)�,如晶型、溶解度���、滲透性�、粒度等�。
(4)分析方法開發(fā)
依據(jù)原料藥質(zhì)量控制項(xiàng)目���,開發(fā)相應(yīng)的分析方法�����,如有關(guān)物質(zhì)���、殘留溶劑���、DNA反應(yīng)性雜質(zhì)�����、元素雜質(zhì)���、含量等�����。殘留溶劑�、元素雜質(zhì)、DNA反應(yīng)性雜質(zhì)�、含量等方法開發(fā)需完全滿足指導(dǎo)原則的要求�。
(5)方法驗(yàn)證
在IND階段�,不需要進(jìn)行全面的方法學(xué)驗(yàn)證�����,但專屬性�����、靈敏度等關(guān)鍵項(xiàng)目是必須驗(yàn)證的項(xiàng)目。由于分析方法隨分析對(duì)象而變化�����,且各具特點(diǎn)�,因此難以確定統(tǒng)一的驗(yàn)證標(biāo)準(zhǔn),通常在IND階段在有充分理由的情況下驗(yàn)證標(biāo)準(zhǔn)可以適當(dāng)放寬。
(6)樣品檢測(cè)與數(shù)據(jù)積累
重點(diǎn)關(guān)注用于安全性研究�����、穩(wěn)定性研究�����、臨床研究等批次樣品的檢測(cè)結(jié)果。
(7)雜質(zhì)譜初步解析
基于工藝條件和現(xiàn)有的知識(shí)對(duì)降解雜質(zhì)�����、樣品中含量超過鑒定限的雜質(zhì)進(jìn)行解析,分析其產(chǎn)生的條件和結(jié)構(gòu)�����。并將研究結(jié)論融入到質(zhì)量控制的過程中�,為雜質(zhì)控制策略的制定提供依據(jù)���。
(8)包裝系統(tǒng)選擇
旨在對(duì)包裝材料選擇提供依據(jù),即影響因素試驗(yàn)(采用內(nèi)包材與采用稱量瓶?jī)煞N形式對(duì)比)或加速�����、長(zhǎng)期穩(wěn)定性試驗(yàn)條件下獲得的重點(diǎn)檢查項(xiàng)目的結(jié)果與0天全檢數(shù)據(jù)對(duì)比�,考察內(nèi)包材選擇是否適宜�。通?����?梢院头€(wěn)定性研究部分合并考察�。
(9)穩(wěn)定性
原料藥穩(wěn)定性包括影響因素(1批供試品)、加速和長(zhǎng)期穩(wěn)定性(3批供試品)�����。穩(wěn)定性考察條件參照ICH Q1A和中國(guó)藥典四部通則9001穩(wěn)定性指導(dǎo)原則設(shè)定(注意需低溫保存的原料藥的考察條件設(shè)置)�。簡(jiǎn)要討論穩(wěn)定性考察相關(guān)的結(jié)果�����、結(jié)論�����,說明擬定的貯藏條件�、復(fù)檢日期或有效期。除注冊(cè)批���、臨床批等樣品的穩(wěn)定性�,還應(yīng)對(duì)毒理批樣品的穩(wěn)定性進(jìn)行考察���,考察時(shí)間至少要覆蓋毒理試驗(yàn)周期。
(10)制定質(zhì)量標(biāo)準(zhǔn)
依據(jù)前期研究數(shù)據(jù)和新藥研究各項(xiàng)指導(dǎo)原則�,初步建立原料藥的質(zhì)量標(biāo)準(zhǔn)并說明質(zhì)量標(biāo)準(zhǔn)的制定依據(jù)。
1.2 制劑質(zhì)量研究
制劑質(zhì)量研究?jī)?nèi)容主要有質(zhì)量控制���、穩(wěn)定性���、包裝和貯存、安慰劑等�����。
(1)控制策略制定
制劑樣品應(yīng)根據(jù)擬選定的劑型和產(chǎn)品特點(diǎn)等來設(shè)置質(zhì)控項(xiàng)目和分析方法�。制劑中雜質(zhì)分為工藝雜質(zhì)�����、降解雜質(zhì)�����、原輔料藥引入雜質(zhì)及外來物遷移(包材相容性雜質(zhì)�、生產(chǎn)組件)。其中含量超過鑒定限的工藝雜質(zhì)和降解雜質(zhì)是研究重點(diǎn)���。與原料藥一樣�,創(chuàng)新藥制劑中一般雜質(zhì)限度參照ICH Q3B制定�����,特定雜質(zhì)制定應(yīng)依據(jù)工藝可行性�����、穩(wěn)定性和產(chǎn)品安全性綜合制定�,尤其要確保雜質(zhì)限度的制定有足夠的安全性數(shù)據(jù)支持�。
(2)分析方法開發(fā)
首先嘗試原料藥分析方法���,如果適用則直接使用同一方法�����,有助于后期雜質(zhì)譜比對(duì)工作�,如果方法不適用,則需要進(jìn)一步優(yōu)化�����。對(duì)于有關(guān)物質(zhì)方法,由于現(xiàn)階段尚處于對(duì)雜質(zhì)安全性的初步研究階段���,對(duì)某些雜質(zhì)可以容忍未達(dá)基線分離甚至合并控制�����。此外在制劑方法開發(fā)過程中注意考察輔料空白對(duì)雜質(zhì)檢測(cè)的干擾���。
(3)分析方法驗(yàn)證
在IND階段�,不需要進(jìn)行全面的方法學(xué)驗(yàn)證,但專屬性�、靈敏度等關(guān)鍵項(xiàng)目是必須驗(yàn)證的項(xiàng)目。由于分析方法隨分析對(duì)象而變化,且各具特點(diǎn)�����,因此難以確定統(tǒng)一的驗(yàn)證標(biāo)準(zhǔn)�����,通常在IND階段在有充分理由的情況下驗(yàn)證標(biāo)準(zhǔn)可以適當(dāng)放寬�����。
(4)結(jié)構(gòu)確證和對(duì)照品標(biāo)化
對(duì)API及雜質(zhì)對(duì)照品進(jìn)行結(jié)構(gòu)確證�����,可引用原料藥相對(duì)應(yīng)的數(shù)據(jù)�。
(5)雜質(zhì)譜初步解析
基于工藝條件和現(xiàn)有的知識(shí)對(duì)降解雜質(zhì)、樣品中含量超過鑒定限的雜質(zhì)進(jìn)行解析�����,分析其產(chǎn)生的條件和結(jié)構(gòu)�����,并將研究結(jié)論融入到質(zhì)量控制的過程中�����,為雜質(zhì)控制策略的制定提供依據(jù)。
(6)穩(wěn)定性研究
在不采用括號(hào)法和矩陣設(shè)計(jì)的情況下�����,每一種規(guī)格和包裝規(guī)格的制劑都應(yīng)進(jìn)行穩(wěn)定性研究。制劑穩(wěn)定性包括影響因素(1批供試品)�����、加速和長(zhǎng)期穩(wěn)定性(3批供試品)���。穩(wěn)定性考察條件參照ICH Q1B和中國(guó)藥典四部通則9001穩(wěn)定性指導(dǎo)原則設(shè)定�。注意需低溫保存的原料藥的考察條件設(shè)置以及產(chǎn)品使用中穩(wěn)定性考察���。簡(jiǎn)要討論穩(wěn)定性考察相關(guān)的結(jié)果�����、結(jié)論,說明擬定的貯藏條件�、復(fù)檢日期或有效期。除注冊(cè)批���、臨床批等樣品的穩(wěn)定性�,還應(yīng)對(duì)毒理批樣品的穩(wěn)定性進(jìn)行考察�����,考察時(shí)間至少要覆蓋毒理試驗(yàn)周期�����。確保穩(wěn)定性數(shù)據(jù)能夠支持在計(jì)劃的臨床研究期間所研制的新藥符合初步擬定的質(zhì)量標(biāo)準(zhǔn)要求���。
(7)安慰劑研究
安慰劑也叫模擬藥物�,是在制劑的基礎(chǔ)上去除原料藥而成�����。安慰劑沒有藥效,也沒有毒副作用�����,常用作藥理試驗(yàn)的對(duì)照品���。安慰劑也需進(jìn)行全面的質(zhì)量控制�,并與制劑在相同的條件下穩(wěn)定性研究�。
(8)樣品檢測(cè)與數(shù)據(jù)積累。
重點(diǎn)關(guān)注用于安全性研究�、穩(wěn)定性研究�����、臨床研究等批次樣品的檢測(cè)結(jié)果���。
(9)制定質(zhì)量標(biāo)準(zhǔn)
依據(jù)前期研究數(shù)據(jù)和新藥各項(xiàng)指導(dǎo)原則�,初步建立制劑的質(zhì)量標(biāo)準(zhǔn)并說明質(zhì)量標(biāo)準(zhǔn)的制定依據(jù)�����。
創(chuàng)新藥研發(fā)具有漸進(jìn)性和不確定性�����,研究工作應(yīng)遵循其規(guī)律開展�����。在IND階段,安全性是重點(diǎn)考量的問題�����,質(zhì)量研究工作也應(yīng)以支持安全性為目的,在合規(guī)的大前提下���,依據(jù)所研發(fā)品種的特征���,科學(xué)合理的制定研究方案,以確保I期臨床期間受試者的安全性。