摘要
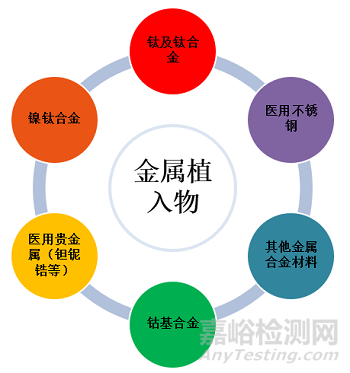
本文以鎳鈦合金心血管植入物為例,深入挖掘植入醫(yī)療器械產品金屬離子析出試驗研究的技術重點����。
對鎳鈦合金心血管植入物進行金屬離子析出試驗研究,主要包含:毒理學允許限量的建立�����,浸提條件和時間的確認��,方法學驗證���,離子析出含量的測定以及毒理學風險評估�。
1���、研究背景
人體是由化學元素組成的,組成人體的元素有60多種�����。人體過量攝入金屬離子可能會導致神經損傷���,呼吸紊亂�����,癌癥發(fā)病率升高等����。金屬植入醫(yī)療器械在人體內由于磨損、腐蝕等原因會導致金屬離子緩慢釋放����。因此植入物植入人體后,離子釋放速率和釋放量是評價其臨床使用安全性的重要內容����。
基于《介入類醫(yī)療器械產品化學性能要求的說明》技術要求和指導原則,鎳鈦合金心血管植入物植入人體后���,鎳鈦合金的鎳離子釋放速率和釋放量是評級其臨床使用安全性的重要內容����。
由于存在體內測試時間長�����、成本高�,測試結果準確率低(機體對實驗結果的干擾較大)等因素��,在臨床前評價階段�����,宜通過建立鎳離子體外釋放模型���,形成鎳離子釋放評價的體外試驗方法,可以更方便評估離子釋放風險����,更好地保證相關產品臨床使用安全性。在臨床前評價階段����,哪些金屬植入物產品需要進行金屬離子析出研究呢?
2�、研究方法
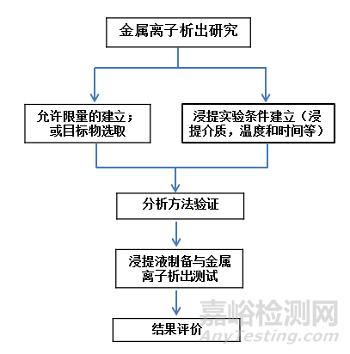
金屬離子析出研究和毒理學風險評估依據(jù)ISO10993-1、ISO 10993-15����、ASTM F3306��、YY/T 1823-2022 《心血管植入物 鎳鈦合金鎳離子釋放試驗方法》和YY/T 1802-2021《增材制造醫(yī)療產品 3D打印鈦合金植入物金屬離子析出評價方法》等進行�,項目研究流程圖如下:
3����、植入物金屬離子析出試驗研究實例
3.1 允許限量(AL) 的建立
依據(jù)ISO 10993-17和YY/T 1823-2022 附錄A等毒理學評估方法����,以鎳鈦合金支架產品為例,進行產品允許限量的建立��。
3.1.1 分析步驟



其中:
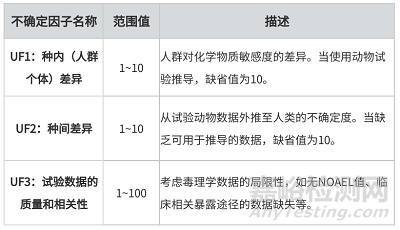
NO[A]EL:No Observed [Adverse] Effect Level�,無可見[有害]作用水平;
LO[A]EL:Lowest Observed [Adverse] Effect Level�����,最小可見[有害]作用水平����;
MF:Modified Factor,即修正因子����;
POD:Point of Departure,關鍵評估終點�����。
3.1.2 推導和計算過程
其中:
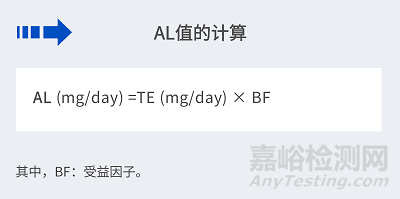
TI ( Tolerable intake):可耐受攝入量����;
TE (Tolerable exposure): 可耐受接觸量�;
AL (Allowable limit): 允許限量�。
表1 不確定因子UF1~UF3描述


基于以上推導和計算過程����,以及結合鎳鈦合金支架產品的臨床使用情況����,建立鎳鈦合金支架產品中Ni元素允許限量。
3.2 浸提實驗條件建立
依據(jù)鎳鈦合金支架產品的接觸風險���,對樣品整體浸泡���,同時進行平行試樣、空白對照試樣和陽性對照試樣制備���。主要的浸提條件如下:
1)浸提樣品:鎳鈦合金支架
2)浸提介質:PBS緩沖溶液(pH=7.4)
3)浸提比例:1c㎡/ml
4)浸提溫度:37±1℃
5)取樣時間點:1d��、2d…60d
6)振蕩頻率:72±2rpm
3.3 金屬離子析出量綜合評價
通過ICP-MS 設備進行Ni 元素測試的方法學設計和開發(fā)驗證�,主要研究內容包括專屬性����、線性、檢出限��、 定量限�、準確度、精密度(重復性和中間精密度)及耐用性����。
通過使用已驗證的方法對Ni金屬離子析出量進行測試,從而獲得Ni金屬離子的階段釋放量�,即Ni離子析出結果,進而進行毒理學風險評估����。根據(jù)Ni離子析出結果綜合評估鎳鈦合金支架產品在不同階段的估計暴露量。即Ni元素前1天釋放量小于mdev,1d����;前30天釋放量小于mdev, 30d;前60天釋放量小于mdev, 60d���;因此�,在鎳鈦合金支架產品中�,Ni元素的毒理學風險被認為是可接受的���。
4、總結與展望
為了更好地保證相關產品臨床使用安全性�����,金屬植入物的離子析出評價將是大勢所趨�����,也是國內審查要點之一����。
本文以鎳鈦合金支架產品為例,依據(jù)YY/T 1823-2022 《心血管植入物鎳鈦合金鎳離子釋放試驗方法》對產品中Ni離子析出評價方法進行了梳理�。該標準主要是針對鎳鈦合金心血管植入物,包括血管支架���、心臟封堵器����、腔靜脈濾器�、心臟瓣膜等等產品。總的來說是通過金屬離子體外釋放模型����,通過Ni元素的允許限量(AL)與估計暴露量進行比較,綜合評估產品的毒理學風險是否可接受�。
目前針對金屬植入物的離子析出評價標準��,主要是YY/T 1802和YY/T 1823�,但這兩個標準的適用范圍相對整個醫(yī)用鈦合金材料及其器械產品來說還是比較窄,目前國際參考ASTM F3306已經推行多年�,但國內金屬離子析出評價方法還未出現(xiàn)具備整體指導作用的通用標準,因而對于金屬離子尤其是人體必需元素的安全性評價還有較長的路要走���。
【參考文獻】
[1] ISO 10993-1, Biological evaluation of medical devices Part 1: Evaluation and testing within a risk management process (2018).
[2] ISO 10993-12, Biological evaluation of medical devices-Part 12: Sample preparation and reference materials (2012).
[3] ISO 10993-15:2019����,Biological evaluation of medical devices — Part 15: Identification and quantification of degradation products from metals and alloys
[4] ISO 10993-17, Biological evaluation of medical devices Part 17: Establishment of allowable limits for leachable substances (2002)
[5] ISO 10993-18, Biological evaluation of medical devices-Part 18: Chemical characterization of medical device materials within a risk management process (2020).
[6] ICH Q2 (R2) Validation of analytical procedures: Text and Methodology.
[7] ICH Q3D(R2), Guideline for elemental impurities.
[8] ChP<0412>, Inductively Coupled Plasma Mass Spectrometry (2020).
[9] ChP<9101>, Guideline for analytical methods of pharmaceutical quality standards (2020).
[10] USP <232> Element Impurities-Limits
[11] USP<233>Element Impurities-Procedure
[12] ASTM F3306-19 Standard Test Method for Ion Release Evaluation of Medical Implants.
[13] YY/T 1802-2021《增材制造醫(yī)療產品 3D打印鈦合金植入物金屬離子析出評價方法》
[14] YY/T 1823-2022《心血管植入物 鎳鈦合金鎳離子釋放試驗方法》
[15] T/CSBM 0011-2021 醫(yī)用鈦合金植入物金屬離子析出評價方法