摘要
藥品研發(fā)數(shù)據(jù)是支持藥品審評對藥品安全性����、有效性��、質量可控評價的重要證據(jù)�����,是藥品注冊現(xiàn)場核查的重要內容��。2020年7月1日《藥品注冊管理辦法》正式生效后,藥品注冊核查也發(fā)生了新的變化���。本文對近2年化學藥品藥學研制現(xiàn)場核查情況進行梳理���,對研制現(xiàn)場核查要點的變化和常見問題進行分析,為新法實施后藥品注冊申請人加強藥品研發(fā)管理提供思路�。
藥品研發(fā)數(shù)據(jù)是支持藥品審評對藥品安全性、有效性����、質量可控評價的重要證據(jù),是藥品注冊現(xiàn)場核查的重要內容[1-3]����?�!端幤饭芾矸ā?、《藥品注冊管理辦法》的修訂與實施,賦予了藥品注冊核查新的定位與要求�。2021年國家藥品監(jiān)督管理局食品藥品審核查驗中心(以下簡稱核查中心)發(fā)布了《藥品注冊核查工作程序(試行)》及《藥品注冊核查要點與判定原則(藥學研制和生產(chǎn)現(xiàn)場)(試行)》,與2008年發(fā)布的《藥品注冊現(xiàn)場核查管理規(guī)定》相比����,進一步強化了藥品注冊核查與審評工作的銜接,進一步細化了藥學研制和生產(chǎn)現(xiàn)場核查的要點[4-8]。本文對近2年化學藥品藥學研制現(xiàn)場核查情況進行梳理���,分析了研制現(xiàn)場核查要點的變化和常見問題��,為新法實施后藥品注冊申請人加強藥品研發(fā)管理提供思路���。
1、藥學研制現(xiàn)場核查要點
2021年發(fā)布的《藥品注冊核查要點與判定原則(藥學研制和生產(chǎn)現(xiàn)場)(試行)》藥學研制現(xiàn)場核查要點(以下簡稱2021年核查要點)包括質量管理��、處方工藝�����、樣品試制����、原輔料與直接接觸藥品的包裝材料和容器(以下簡稱原輔包)、質量控制����、對照品和參比制劑、穩(wěn)定性���、數(shù)據(jù)可靠性9個方面�����,它在2008年《藥品注冊現(xiàn)場核查管理規(guī)定》藥品注冊現(xiàn)場核查要點及判定原則藥學方面(以下簡稱2008年核查要點)工藝及處方研究(1.1~1.3)����,樣品試制(2.1~2.8),質量���、穩(wěn)定性研究及樣品檢驗(3.1~3.10)����,委托研究(4)的基礎上�����,結合了我國藥品研發(fā)實際和藥品監(jiān)管工作需要�,充分吸收了ICH,WHO��,PIC/S等國際通行的指南內容����,也更加貼合申報資料��。相關核查要點對應關系見表1。

2021年核查要點在如下幾個方面進行了深化:①將質量管理作為一個專項進行描述�,包括了組織機構與人員、研究記錄���、文件和記錄�、變更和偏差管理���、委托研究5個方面的要求����。2021年核查要點既包含了2008年核查要點中關于與研發(fā)工作相適應的儀器設備�����、場所����,委托研究的相關要求,還進一步提出了建立與研究內容相適應的組織機構和質量管理體系���,制訂相應的管理制度或標準操作規(guī)程并遵照實施����,并強調了藥物進入臨床階段后的變更和偏差管理。②新增技術轉移核查要點�,將其看作一個系統(tǒng)工程,關注研制過程中所獲取的產(chǎn)品知識和經(jīng)驗是否轉移給生產(chǎn)企業(yè)����,接受技術轉移的生產(chǎn)企業(yè)是否有能力實施被轉移的技術、生產(chǎn)出符合注冊要求的藥品���。③從申報資料出發(fā)���,將2008年核查要點樣品試制,質量��、穩(wěn)定性研究及樣品檢驗��,深化為2021年核查要點中樣品試制�、原輔包,質量控制����、對照品、穩(wěn)定性����,同時增加了參比制劑的相關要求,明確關鍵批次樣品在上市申請批準前不得銷毀��,強調了原輔包內控標準的制定����、穩(wěn)定性方案的制定與實施等。④與時俱進在2008年核查要點關于申報資料真實����、一致、可追溯的基礎上����,進一步明確研制單位應當采取有效措施防止數(shù)據(jù)的修改、刪除���、覆蓋等�����,以確保數(shù)據(jù)可靠?��,F(xiàn)行的核查要點是落實深化藥品審評審批改革,鼓勵藥品創(chuàng)新政策的具體體現(xiàn)���。
2�、近2年化學藥品藥學研制現(xiàn)場核查概況
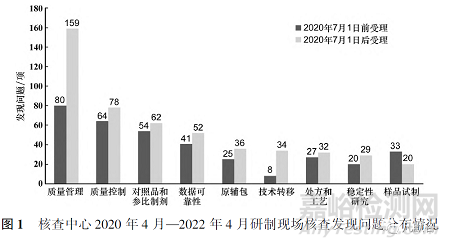
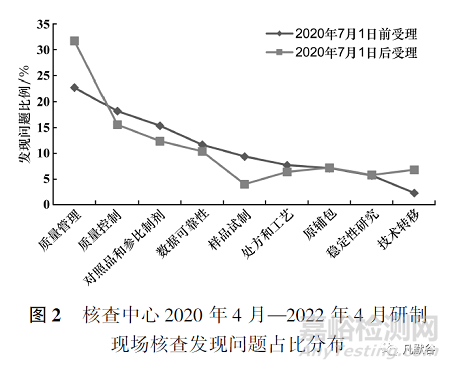
本文對核查中心2020年4月—2022年4月組織的研制現(xiàn)場核查進行分析,篩選了189個化學藥品的藥學研制現(xiàn)場核查�,其中2020年7月1日前按照《總局關于調整藥品注冊受理工作的公告》(2017年第134號)集中受理品種83個,核查標準為2008年核查要點�,共發(fā)現(xiàn)問題352項,平均每個品種發(fā)現(xiàn)問題4.2項����;2020年7月1日后按2020年《藥品注冊管理辦法》受理的品種109個,核查標準為2021年核查要點����,發(fā)現(xiàn)問題502項,平均每個品種發(fā)現(xiàn)問題4.6項��。本文將189個藥學研制現(xiàn)場核查發(fā)現(xiàn)問題按照表1對應關系進行重新統(tǒng)計分析�����,發(fā)現(xiàn)質量管理方面的問題最為突出�����,其次是質量控制����、對照品和參比制劑����、數(shù)據(jù)可靠性方面的缺陷��。對于2020年7月1日后受理品種質量管理����、技術轉移兩方面的發(fā)現(xiàn)問題較2020年7月1日受理前的品種有明顯的增長趨勢����。見圖1和圖2。
3����、藥學研制現(xiàn)場核查常見問題分析
3.1質量管理
從近2年的核查發(fā)現(xiàn)問題分析,質量管理方面的問題最為突出����,主要包括:①制度不健全,包括缺少關鍵性質量管理文件����,缺少技術轉移���、質量標準管理、實驗記錄與數(shù)據(jù)管理等文件���;文件缺乏適用性�,文件規(guī)定與現(xiàn)行法規(guī)要求不符���;文件內容規(guī)定不詳���,缺少具體的流程和操作要求。②人員培訓不到位����,相關人員不熟悉操作流程或管理要求,同類問題反復發(fā)生�。③文件規(guī)定和實際執(zhí)行不一致。未按要求對記錄進行受控管理�,隨意發(fā)放、使用���,無受控措施��;記錄不完整�����,缺少實驗儀器/試劑/色譜柱來源信息�、缺少部分制備過程及參數(shù)、缺少數(shù)據(jù)處理方式等���;未按程序進行偏差管理,如中間體收率及含量不符合要求���、不同亞批之間中間體波動超出規(guī)定范圍�、缺少調查評估���,樣品結果異常���、超趨勢等直接復測,不合格檢測結果未按文件規(guī)定雙人復核�����,穩(wěn)定性考察過程中光照度及溫濕度等不符合要求���、不溶性微粒檢測用水不符合《中華人民共和國藥典》要求���、未按程序調查等�;未按程序進行變更管理如部分方法���、物料變更未說明變更原因或依據(jù)�����,變更生效后相關的文件�����、標準����、方案未按要求修訂����。④委托研究管理不到位。如未對委托方的能力�����、質量體系進行評估;委托研究結果審核不嚴���,研究結果與記錄或附圖不一致�����;文件和樣品交接不可追溯等�。
3.2質量控制
質量研究相關的分析方法學驗證���、質量對比��、樣品檢驗、儀器設備的檢定或校驗等一直是核查的重要內容��。常見問題如下:①申報資料中部分數(shù)據(jù)與原始記錄不一致����,包括申報資料中考察時間,檢測方法或條件�、樣品處理過程等與原始記錄不一致。②分析方法學驗證不規(guī)范�,如中間精密度實驗由同一位分析員在不同日期完成;分析方法驗證方案和報告未按照企業(yè)程序進行審核�。③檢驗報告不規(guī)范,檢驗依據(jù)缺乏追溯性,引用廠家報告或自委托檢驗結果的項目缺少標注/說明��。④儀器設備校驗不規(guī)范�。如滲透壓儀、刻度吸管�����、移液槍等未校驗或驗證范圍未涵蓋實際使用范圍��;自校的標準器具精度不滿足要求���,校驗條件或限度不滿足檢驗規(guī)程要求����;校驗操作規(guī)程生效時間晚于開始校驗的時間�����;校驗記錄無法追溯所使用的驗證工具��、標準品����、校驗部件��;校驗報告無結論����;自校后未規(guī)定使用有效期���。⑤儀器設備使用不規(guī)范��。如部分儀器未完成驗證即開始使用����,未根據(jù)驗證結果制定使用要求�,儀器設備故障維修后未評估或再確認/再驗證。
3.3對照品和參比制劑
對照品和參比制劑方面最常見的問題有:①未按要求貯存和使用����。如對照品未按要求使用惰性氣體保存����、參比制劑未按照規(guī)定的貯存溫度保存、對照品未按要求使用前干燥至恒重����,要求單次使用的對照品實際多次開瓶使用��,對照品或配制的儲備液未規(guī)定有效期�、有效期制定無數(shù)據(jù)支持或超效期使用�,使用記錄填寫不完整、描述模糊���,不便追溯來源和用途���。②對照品未按要求進行標定。如文件規(guī)定對照品應標定后使用或實際直接采用原料藥成品報告書中結果����,文件規(guī)定雙人標定或采用2種方法進行標定,實際一人標定或采用一種方法標定后即使用�����。③參比制劑使用時間早于入庫時間�,參比制劑入賬數(shù)量少于購買數(shù)量未記錄原因或保留憑證。
3.4數(shù)據(jù)可靠性
數(shù)據(jù)可靠性的主要問題包括申報資料相關內容無法溯源���,電子數(shù)據(jù)可靠性保障不足�、選擇性使用數(shù)據(jù)以及支持結論的部分信息未在申報資料中描述����。包括:①紙質�、電子數(shù)據(jù)無法追溯���。如現(xiàn)場未能提供部分研究或檢驗項目的原始記錄�,缺少部分批次電子原始數(shù)據(jù)或提供的電子圖譜無法讀取��。②電子數(shù)據(jù)可靠性保障不足����。如關鍵批次在無審計追蹤功能的儀器上檢測,關鍵批次生產(chǎn)或檢驗時儀器設備審計追蹤未開啟或開啟不全�����,用戶登錄不受控�����,實驗相關人員刪除��、覆蓋�����、手動積分等權限不受控�����,部分儀器設備時間不受控可隨意更改���。③電子數(shù)據(jù)保存不規(guī)范�。如電子圖譜缺失批號���、名稱���、存儲路徑等追溯性信息,部分儀器設備電子數(shù)據(jù)未備份�、歸檔,歸檔的電子數(shù)據(jù)不全或缺少相關的審計日志等��。④多次實驗或檢驗���,僅記錄或計算部分結果����。如使用24片多次開展溶出度實驗����,選取第1次實驗中合格的6片和第3次檢測的6片數(shù)據(jù)作為溶出度數(shù)據(jù)�����;儀器中有3次實驗����,原始記錄中僅有第1次和最后1次�。⑤影響結果判斷的信息不全。如申報資料中僅描述樣品的考察時間和條件���,未描述樣品的生產(chǎn)日期�,質量對比實驗結論為自制制劑優(yōu)于參比制劑��;申報資料中變更信息描述不全����,如部分批次的生產(chǎn)設備、工藝參數(shù)����,部分項目的檢驗方法、樣品處理方法等��。
3.5技術轉移
從發(fā)現(xiàn)問題來看����,主要為技術轉移程序執(zhí)行不嚴,轉移方案內容不全����,包括:①未按規(guī)定方式轉移。如未規(guī)定的形式和流程實施工藝����、分析方法轉移工作,缺少正式的轉移文件和記錄或未按規(guī)定的程序進行雙方確認或審批����;轉移文件歸檔不全。②技術轉移方案或計劃內容不全�。如未明確接受標準,未對變更事項�、人員、設備�、工藝、物料等因素進行評估����,未說明輔料型號����、對照品�、色譜柱及其他特殊要求。③技術轉移過程不規(guī)范�����。轉移過程中的生產(chǎn)偏差�,分析方法轉移中出現(xiàn)的異常情況,未進行分析評估�����。④轉移內容不完整�。未進行分析方法的轉移確認;轉移后轉出方對生產(chǎn)工藝����、分析方法的再研究或優(yōu)化未及時轉移給接收方。
4���、探討與建議
研制現(xiàn)場核查是基于申報資料�����,通過原始記錄和數(shù)據(jù)����、藥品研制合規(guī)性����、數(shù)據(jù)可靠性,確認申報資料真實性��、一致性的過程�����。近2年研制現(xiàn)場核查中發(fā)現(xiàn)質量管理�、質量控制、數(shù)據(jù)可靠性�����、技術轉移是研制現(xiàn)場核查的主要問題���。進一步分析各問題都不是孤立存在的����,關鍵在于藥品研發(fā)階段是否建立了成熟的質量管理體系,進而實現(xiàn)研發(fā)與生產(chǎn)的有機銜接�����。如電子和紙質記錄的清晰��、準確����、及時、原始����、可追溯,保持受控狀態(tài)�����,既是質量管理的重要內容���,也是數(shù)據(jù)可靠性的保障之一�。技術轉移要求將在研制過程中所獲取的控制工藝及文件和專業(yè)知識轉移給生產(chǎn)企業(yè)[9]�����。技術轉移前的評估、變更及注意事項�����、明確的接受標準���、完整的轉移方案、轉移過程的執(zhí)行和偏差處理以及最終的總結���,既需要制度明確職責�、流程和要求����,也需要具體在實踐中檢驗接收是否理解、掌握了品種相關的知識[10]�����。藥品研制單位應重視2021年核查要點中的新要求新變化�����,將其放在國內外法規(guī)指南變化的大環(huán)境中去理解,結合研制現(xiàn)場常見問題和企業(yè)實際情況�,通過自查確定當前藥學研制的工作中的不足,以完善質量管理����,在實踐中持續(xù)改進藥品研制的管控要求,減少研制過程中的相關問題和風險�����。
藥學研制工作質量直接影響藥品能否上市及上市速度�����,該階段的研究工作是證明藥品質量的關鍵證據(jù)�����。通過本分析研究���,希望為制藥行業(yè)進一步加強藥學研究工作�����,做好藥學研制現(xiàn)場核查準備提供參考����。

