前言
創(chuàng)新藥,也稱為原研藥����,是一個相對于仿制藥的概念,指的是從機(jī)理開始源頭研發(fā)�,具有自主知識產(chǎn)權(quán),具備完整充分的安全性有效性數(shù)據(jù)作為上市依據(jù)����,首次獲準(zhǔn)上市的藥物。按照藥物研發(fā)的常規(guī)流程����,一款藥物從確定靶點(diǎn)到最后審批上市的整個研發(fā)周期通常耗時十?dāng)?shù)年的時間�。專利保護(hù)期過后,就會被大量仿制��。在美國食品藥品監(jiān)督管理局(FDA)的評審標(biāo)準(zhǔn)中�,新分子實(shí)體(New Moleculer Entity,NME)和獲得新生物制品許可的藥物(Biologic License Application�,BLA)均屬于創(chuàng)新藥。根據(jù)CFDA頒布的注冊分類����,分為五大類。本文中的新藥特指1類新藥。
新藥研發(fā)模式
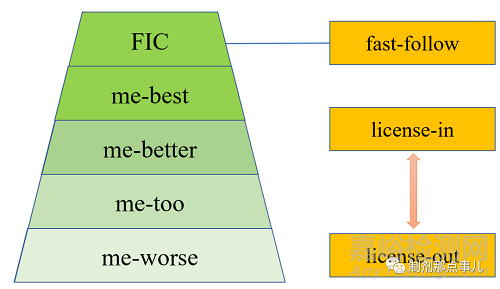
1類新藥大致可分為以下幾類(圖1)�。
▲ 圖1-新藥研發(fā)模式
“First in Class”即為首創(chuàng)新藥。一般指能治療某種疾病的新靶點(diǎn)����、新機(jī)理藥物;
“me-too”意為與FIC具有相似的作用機(jī)理和治療效果的藥物����,;
“me-better”結(jié)構(gòu)可能具有較大優(yōu)化��,在活性����、代謝和/或毒性等各方面優(yōu)于"me-too"藥;
“me-best”即在同類創(chuàng)新藥里最好����;
“me-worse”則是相比“me-too”而言在療效更差或毒副作用更強(qiáng)。
“fast-follow”指快速追蹤新藥的模式�,以最快速度對“first in class”藥物分子進(jìn)行改造,也被稱為“站在巨人的肩膀上依葫蘆畫瓢”����;
“license-in”指購買授權(quán)許可����;
“license-out”指出售授權(quán)許可��。
藥物發(fā)現(xiàn)階段
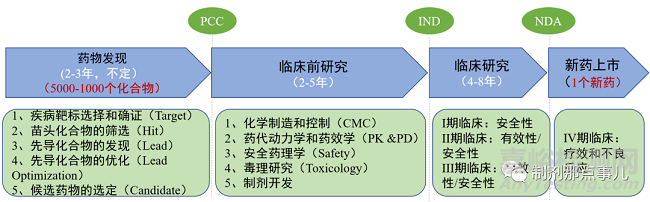
在生物醫(yī)藥研發(fā)領(lǐng)域�,新藥研發(fā)具有長周期、高成本��、高風(fēng)險的特點(diǎn)�。業(yè)內(nèi)人常說的“雙十定律”即成功研發(fā)一款新藥,即使過程比較順利����,也需要耗時十年、投資十億美金����,甚至更久更多��。
1�、疾病靶標(biāo)選擇與確證(Target)。
2�、苗頭化合物的篩選(Hit)。
3�、先導(dǎo)化合物的發(fā)現(xiàn)(Lead)��。
4��、先導(dǎo)化合物的優(yōu)化(Lead Optimization)�。
▲ 圖2-新藥開發(fā)基本流程
IND申報及審批
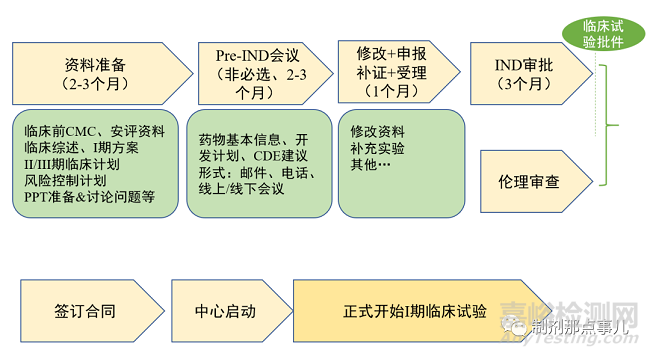
在候選藥物完成臨床前研究后��,便可向藥品評審中心(Center for Drug Evaluation,CDE)(美國為FDA)提出臨床試驗(yàn)申請(Investigational New Drug����,IND)一般以IND作為新藥研發(fā)的第二個關(guān)鍵節(jié)點(diǎn)。
1����、資料準(zhǔn)備階段。包括臨床前研究的質(zhì)量研究和安全性評價資料����、臨床試驗(yàn)方案、藥效學(xué)/藥代動力學(xué)/毒理學(xué)資料等(圖3)��。
▲ 圖3-IND申報流程
2����、Pre-IND會議。在正式向CDE提交IND之前�,申請人可自愿選擇是否進(jìn)行Pre-IND會議(無償)����,向監(jiān)管機(jī)構(gòu)提交產(chǎn)品的基本信息�、開發(fā)計劃,所需溝通交流問題的背景和數(shù)據(jù)�,并提出擬解決問題的建議和想法等。對于缺乏新藥注冊的企業(yè)來說��,這是保證成功的必要一步�。
3、IND審批�。CDE會在60天內(nèi)通知申請人該新藥物種是否可進(jìn)行臨床試驗(yàn),若60天內(nèi)未給出意見�,則默認(rèn)可以開始臨床試驗(yàn)。
4��、倫理審查����。
5、簽訂合同�。
6�、中心啟動會。臨床試驗(yàn)正式開始前�,必須在研究科室召開臨床試驗(yàn)啟動會��,其目的了讓所有參與臨床試驗(yàn)的人員熟悉試驗(yàn)方案及具體操作流程��。參與人員主要包括:研究者��、申辦方��、機(jī)構(gòu)人員�、CRA和CRC等��。經(jīng)過啟動會的當(dāng)天準(zhǔn)備�、召開,待啟動會結(jié)束后����,即可正式開展I期臨床試驗(yàn)。
藥物臨床試驗(yàn)
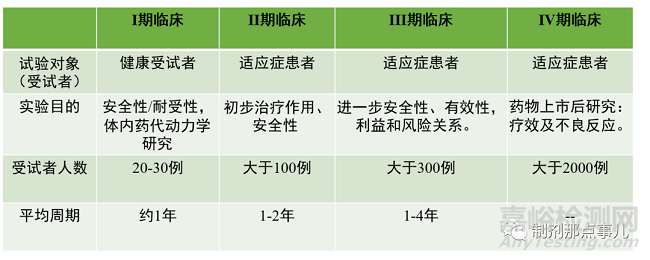
藥物臨床試驗(yàn)共分為四期����,其中,I-III期臨床試驗(yàn)為上市前��,且每一階段臨床試驗(yàn)都會預(yù)設(shè)臨床終點(diǎn)����,達(dá)到臨床終點(diǎn)方可開展下一階段的試驗(yàn)����;IV期臨床試驗(yàn)為上市后研究�。
▲ 圖4-藥物臨床試驗(yàn)
1、I期臨床試驗(yàn)����。受試者為健康人。主要目的是:1)對藥物的安全性及在人體的耐受性進(jìn)行研究�,考察藥物副作用與藥物劑量遞增之間的關(guān)系;2)考察藥物的體內(nèi)藥代動力學(xué)性質(zhì)����,包括代謝產(chǎn)物及代謝途徑等。按照試驗(yàn)階段可分為Ia期和Ib期�。Ia期也稱為單次劑量遞增(SAD,Single ascending dose)試驗(yàn),也是我們常說的“爬坡試驗(yàn)”����。分析關(guān)鍵數(shù)據(jù)指標(biāo)結(jié)果,直到達(dá)到預(yù)期水平�。為制定接下來II、III期臨床試驗(yàn)設(shè)計和給藥方案提供依據(jù)����。若藥物增加新適應(yīng)癥�,一般不需要再做I期臨床試驗(yàn)����。
2����、II期臨床試驗(yàn)。受試者為適應(yīng)癥患者����。重點(diǎn)在于初步評價藥物的安全性和療效。應(yīng)用安慰劑或已上市藥物作為對照藥物對新藥的療效進(jìn)行評價�,在此過程中對疾病的發(fā)生發(fā)展過程對藥物療效的影響進(jìn)行研究;確定III期臨床試驗(yàn)的給藥劑量和方案��;獲得更多的藥物安全性方面的資料��,按階段可分為IIa期和IIb期����。IIa期(Proof of Concept,POC)即療效探索����,目的是證明藥物的臨床療效和生物活性��。這個階段通常用于少數(shù)患者��,主要療效終點(diǎn)在給藥后早期��,評估其有效性��;IIb期(Dose Finding��,DF)即劑量確定����,目的是確定顯示生物活性�,而副作用最小的最佳劑量,在這個階段評估藥物的療效以及安全性�,為了找到III期試驗(yàn)中最佳劑量,主要療效終點(diǎn)在給藥后后期����。
3、III期臨床試驗(yàn)�。受試者為適應(yīng)癥患者,也被稱為治療作用確證(confirmatory)�。目的進(jìn)一步更大范圍驗(yàn)證藥物安全性和療效�,對藥物的益處/風(fēng)險進(jìn)行評估����。III期試驗(yàn)是最昂貴,最耗時和最困難的��,試驗(yàn)設(shè)計通常是隨機(jī)����、對照����、多中心的?���?煞譃镮IIa期和IIIb期。IIIa期試驗(yàn)是在藥物的有效性被證明后����,但在向監(jiān)管機(jī)構(gòu)提交注冊申請之前進(jìn)行的。研究的結(jié)果用于提交新藥注冊申請�;IIIb期在提交注冊申請后,但在獲得藥品批準(zhǔn)并投入生產(chǎn)之前進(jìn)行����,目的是為了獲得額外的安全性數(shù)據(jù)��、發(fā)表文章����、營銷聲明或準(zhǔn)備藥物上市��。這也被稱為上市前階段(pre-marketing phase)����。
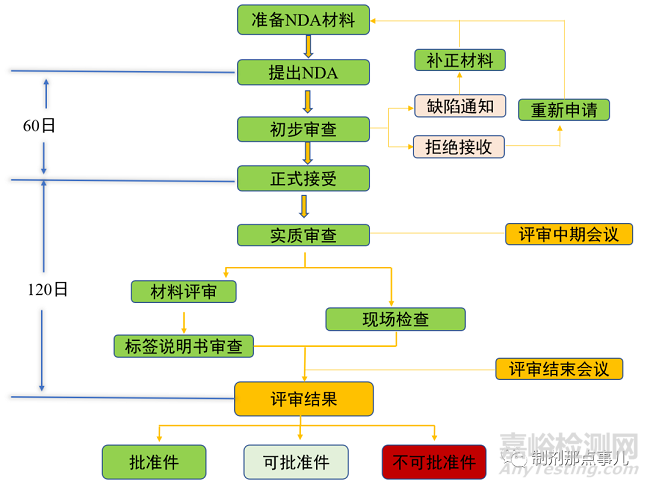
完成III期臨床試驗(yàn)后,便可向國家藥品監(jiān)督管理局(National Medical Products Administration�,NMPA)遞交新藥上市申請(New Drug Application,NDA)受理批準(zhǔn)后便可進(jìn)行新藥上市(圖6)����,一般以NDA作為新藥研發(fā)的第三個關(guān)鍵節(jié)點(diǎn)。
▲ 圖5-新藥NDA流程
新藥上市后
1����、IV期臨床試驗(yàn)。也被稱為上市后研究(PMS�,Post-Marketing Study)或上市后監(jiān)測(PMS,Post Marketing Surveillance)�,主要目的是確定長期的安全性和有效性�?���?梢栽诟L的時間和更大患者群體中對藥物安全性進(jìn)行進(jìn)一步的監(jiān)測和評估。
2�、上市后再審批。一般上市后4-10年����,主要是重新審核NDA中藥物的安全性和有效性��。