隨著新藥研發(fā)技術(shù)的不斷進(jìn)步和審評(píng)制度改革���,創(chuàng)新藥投入臨床使用的速度正在加快�,這是另所有研發(fā)人員振奮鼓舞的事情�,但是一個(gè)全新化合物如此快速投入臨床并應(yīng)用于人,相應(yīng)的風(fēng)險(xiǎn)也隨之增加,這個(gè)風(fēng)險(xiǎn)一方面來(lái)源于化合物本身,一方面來(lái)源于雜質(zhì),因此對(duì)雜質(zhì)進(jìn)行科學(xué)系統(tǒng)的研究,對(duì)于防范和降低臨床風(fēng)險(xiǎn)意義重大。
什么是雜質(zhì)呢?雜質(zhì)是指存在于原料藥中的與原料藥結(jié)構(gòu)不一致的任何成分。藥物在生產(chǎn)����、儲(chǔ)存和運(yùn)輸過(guò)程中都有可能產(chǎn)生雜質(zhì)�。
2����、雜質(zhì)分類
雜質(zhì)的分類方式有很多�,按照理化性質(zhì)可以分為無(wú)機(jī)雜質(zhì)、有機(jī)雜質(zhì)和殘留溶劑�;按照產(chǎn)生途徑可以分為工藝雜質(zhì)和降解雜質(zhì)�;按照其毒性可以分為普通毒性和毒性雜質(zhì);按照其控制策略可以分為特定雜質(zhì)和非特定雜質(zhì)��;按照其產(chǎn)生的風(fēng)險(xiǎn)可以分為潛在雜質(zhì)和實(shí)際存在雜質(zhì)���。
對(duì)雜質(zhì)的分類可以根據(jù)分析目的進(jìn)行選擇��,例如工藝研究總結(jié)中通常需要對(duì)雜質(zhì)產(chǎn)生途徑(降解雜質(zhì)���、工藝雜質(zhì))進(jìn)行描述;在安全評(píng)價(jià)中則需要區(qū)分是普通雜質(zhì)還是毒性雜質(zhì)��,在分析方法選擇時(shí)通常是按照有機(jī)雜質(zhì)(含DNA反應(yīng)性雜質(zhì))��、無(wú)機(jī)雜質(zhì)�����、殘留溶劑來(lái)分類。
3���、雜質(zhì)研究的基本思路
2010年雜質(zhì)譜控制的理念被提出����,使得雜質(zhì)研究由個(gè)別雜質(zhì)控制上升為系統(tǒng)控制����,雜質(zhì)研究的工作也不再是單純的雜質(zhì)檢測(cè)與分析工作,成為了一項(xiàng)需要由工藝���、質(zhì)量��、制劑�、安全性評(píng)價(jià)等多學(xué)科多領(lǐng)域共同完成的系統(tǒng)工程���。
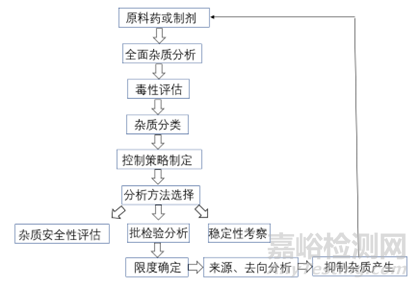
雜質(zhì)譜研究的思路和策略源于QbD理念��,經(jīng)過(guò)多年的摸索和研究����,已經(jīng)從以終為始的被動(dòng)控制模式轉(zhuǎn)化為以源為始的主動(dòng)控制模式�����,也就是從雜質(zhì)的來(lái)源入手���,結(jié)合產(chǎn)品的結(jié)構(gòu)特點(diǎn)��、生產(chǎn)工藝�����、處方組成����、存儲(chǔ)條件����、穩(wěn)定性特征等信息全面分析產(chǎn)品中可能存在的雜質(zhì),然后對(duì)雜質(zhì)存在的可能性大小����,是否容易清除、安全等級(jí)進(jìn)行評(píng)估���,建立適宜的分析方法�����,確保有效檢出和確認(rèn)各種潛在雜質(zhì)����;在此基礎(chǔ)之上,結(jié)合生產(chǎn)工藝��、穩(wěn)定性特征�,跟蹤雜質(zhì)在安全性或臨床試驗(yàn)結(jié)果中產(chǎn)生的影響,在符合指導(dǎo)原則的前提下制定每個(gè)特定雜質(zhì)的限度����;最后是對(duì)雜質(zhì)的來(lái)源和去向進(jìn)行分析,通過(guò)工藝控制��、包裝改進(jìn)或儲(chǔ)存條件調(diào)整來(lái)有效抑制雜質(zhì)的產(chǎn)生�����,從源頭上有效把控雜質(zhì)��。
圖1 創(chuàng)新藥雜質(zhì)研究一般思路
本文按照無(wú)機(jī)雜質(zhì)��、殘留溶劑和有機(jī)雜質(zhì)的順序逐一探討各類雜質(zhì)的具體研究思路和控制策略���。
3.1無(wú)機(jī)雜質(zhì)
藥品中的無(wú)機(jī)雜質(zhì)包括殘留的催化劑�����、配體�����、試劑�、重金屬及無(wú)機(jī)鹽等��。對(duì)于無(wú)機(jī)鹽和普通試劑可以參照藥典附錄中的方法進(jìn)行控制����,例如氯化物、硫酸鹽���、熾灼殘?jiān)葯z查項(xiàng)��。在無(wú)機(jī)雜質(zhì)中重點(diǎn)應(yīng)關(guān)注的是元素雜質(zhì)�。
元素雜質(zhì)的來(lái)源包括有意添加的無(wú)機(jī)試劑或催化劑引入��;原輔料中潛在的元素雜質(zhì)�;生產(chǎn)設(shè)備引入及包裝系統(tǒng)中浸出的元素雜質(zhì)�����,依據(jù)風(fēng)險(xiǎn)評(píng)估��,元素雜質(zhì)可以分為4個(gè)風(fēng)險(xiǎn)等級(jí)(I�、2A�����、2B����、3),其中I及2A類元素雜質(zhì)無(wú)論是否有意添加均應(yīng)進(jìn)行風(fēng)險(xiǎn)評(píng)估����,2B類元素雜質(zhì)僅在有意添加時(shí)進(jìn)行風(fēng)險(xiǎn)評(píng)估,3類元素雜質(zhì)��,有意添加時(shí)應(yīng)進(jìn)行風(fēng)險(xiǎn)評(píng)估��,在非有意添加時(shí)依據(jù)所選擇的劑型決定是否進(jìn)行風(fēng)險(xiǎn)評(píng)估����。元素雜質(zhì)的控制策略中���,有一個(gè)“控制閾值”值得關(guān)注,控制閾值是元素水平相當(dāng)于PDE的顯著性水平����,相當(dāng)于PDE 30%,當(dāng)多批既定工藝條件下的樣品檢測(cè)結(jié)果表明所有來(lái)源的某種元素雜質(zhì)的殘留水平均低于控制閾值��,那么����,在對(duì)數(shù)據(jù)進(jìn)行評(píng)估并證明已對(duì)該元素雜質(zhì)進(jìn)行了足夠控制的情況下�,無(wú)需額外控制。否則��,需建立控制方法加以控制����。舉個(gè)例子,比如某口服制劑原料藥的起始物料的生產(chǎn)工藝中使用了金屬催化劑鈀(PDE 100μg/天�,元素允許濃度10μg/g),原則上需要考慮鈀所帶來(lái)的風(fēng)險(xiǎn)��,當(dāng)我們檢驗(yàn)了多批起始物料����、中間體及成品后發(fā)現(xiàn)鈀的殘留量均為未超過(guò)3 μg/g(PDE的30%)�����,那么我們無(wú)需建立控制方法�����。
3.2殘留溶劑
殘留溶劑指原輔料生產(chǎn)或制劑制備過(guò)程中使用或產(chǎn)生的有機(jī)揮發(fā)物����。依據(jù)風(fēng)險(xiǎn)評(píng)估�����,殘留溶劑可以分為4類���,1類溶劑具有不可接受的毒性或危害環(huán)境��,應(yīng)避免使用����;2類溶劑具有非遺傳毒性的動(dòng)物致癌性��,可能導(dǎo)致神經(jīng)毒性、致畸性等不可逆毒性或其他嚴(yán)重但可逆的毒性��,應(yīng)限制使用��;3類溶劑屬于低毒性溶劑���,限度可按照5000ppm或以上來(lái)控制����。4類溶劑屬于沒(méi)有足夠毒理學(xué)數(shù)據(jù)的溶劑�����。其中�����,前3類溶劑在ICH指導(dǎo)原則中均有明確的限度��,第4類溶劑由于缺乏足夠的毒理學(xué)數(shù)據(jù)沒(méi)有明確的限度�。在對(duì)第4類溶劑進(jìn)行研究時(shí)�����,應(yīng)對(duì)毒性數(shù)據(jù)庫(kù)和相關(guān)文獻(xiàn)進(jìn)行全面檢索,將所能獲得的毒性數(shù)據(jù)匯總�����,分別計(jì)算限度(計(jì)算方法參考ICH Q3C)�,然后選擇其中的最低限度作為該溶劑的限度,當(dāng)有新的研究數(shù)據(jù)發(fā)表并且所計(jì)算的限度更低時(shí)應(yīng)及時(shí)對(duì)限度進(jìn)行修訂��。
在對(duì)殘留溶劑進(jìn)行分析時(shí)����,除了實(shí)際使用的溶劑還應(yīng)注意潛在可能產(chǎn)生的溶劑,如同時(shí)使用甲醇和乙酸可能生成乙酸甲酯��、使用甲苯時(shí)需同時(shí)控制其中可能存在的苯等�。
3.3有機(jī)雜質(zhì)
有機(jī)雜質(zhì)主要是指有關(guān)物質(zhì),也就是與生產(chǎn)工藝和藥物結(jié)構(gòu)有關(guān)的雜質(zhì)�。有關(guān)物質(zhì)通常包括:起始物料、中間體���、副產(chǎn)物�����、降解產(chǎn)物等�����。有關(guān)物質(zhì)的控制是藥物質(zhì)量控制的重點(diǎn)內(nèi)容�����,控制的難度也較無(wú)機(jī)雜質(zhì)和殘留溶劑要高�。
按照雜質(zhì)譜研究的一般規(guī)律,有關(guān)物質(zhì)研究首先也是對(duì)原料藥或制劑產(chǎn)品中可能存在的雜質(zhì)進(jìn)行全面的分析�����,列出所有的潛在雜質(zhì)并對(duì)雜質(zhì)產(chǎn)生的可能性大小和毒性進(jìn)行評(píng)估和分類��,1)無(wú)論理論上產(chǎn)生可能性高低��,實(shí)際未檢出或檢出量較低����,且穩(wěn)定性中未見明顯增長(zhǎng)的雜質(zhì)����,可以按照非特定雜質(zhì)來(lái)控制。2)對(duì)于那些實(shí)際檢出量很高或穩(wěn)定性中明顯增長(zhǎng)的雜質(zhì),應(yīng)按照特定雜質(zhì)來(lái)控制�。3)對(duì)那些含有警示結(jié)構(gòu)的雜質(zhì)應(yīng)先按照DNA反應(yīng)性雜質(zhì)進(jìn)行評(píng)估,如在充分研究后排除了其致癌致突變性�,則可按照一般雜質(zhì)來(lái)控制,否則列入DNA反應(yīng)性雜質(zhì)控制隊(duì)伍之中����。
在確定初步的研究策略后我們?cè)賮?lái)看下各種雜質(zhì)限度如何制定。
3.3.1 普通雜質(zhì)
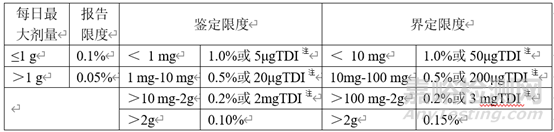
ICH Q3A和Q3B中分別給出了原料藥和制劑產(chǎn)品中雜質(zhì)限度匯總表��。
表1 原料藥雜質(zhì)限度
表2 制劑雜質(zhì)限度
注:取限度低者
對(duì)于創(chuàng)新藥�,非特定雜質(zhì)(即單一最大雜質(zhì))可按照上表中的限度進(jìn)行控制,而特定雜質(zhì)在有充足數(shù)據(jù)支撐的前提下可以超越指導(dǎo)原則中的限度要求��,這是因?yàn)閯?chuàng)新藥在申請(qǐng)臨床研究之前開展大量的毒理學(xué)研究��,那些含量超過(guò)鑒定限或界定限的雜質(zhì)以及穩(wěn)定性性研究中明顯增長(zhǎng)至超過(guò)鑒定限或界定限的雜質(zhì)都可以在毒理學(xué)研究中得到安全性評(píng)價(jià)�,所獲得數(shù)據(jù)可以用于支持相應(yīng)雜質(zhì)限度的制定。
舉個(gè)例子:
1)某原料藥中雜質(zhì)A含量為0.13%�,該批原料藥用于犬長(zhǎng)毒實(shí)驗(yàn),NOAEL值為40 mg/kg/天����。假設(shè)臨床劑量為150mg.d-1,以患者體重60kg計(jì)����,則人的給藥劑量為2.5mg.kg-1.d-1�,與犬的給藥劑量比較��,可支持該雜質(zhì)的安全限度應(yīng)為(40/2.5)*0.13%=2.08%
2)6批一定規(guī)模樣品中雜質(zhì)A平均含量0.13%���,標(biāo)準(zhǔn)偏差0.04%��,該雜質(zhì)限度0.25%(平均值+3 SD)��。
3)3批上述原料藥長(zhǎng)期放樣24個(gè)月�����,雜質(zhì)A含量最高增長(zhǎng)至0.56%���,依據(jù)穩(wěn)定性結(jié)果該雜質(zhì)限度為0.6%。
綜上�,依據(jù)安全性、工藝可行性及穩(wěn)定性的結(jié)果綜合制定該雜質(zhì)的限度可定為0.60%�。
3.3.2 DNA反應(yīng)性雜質(zhì)
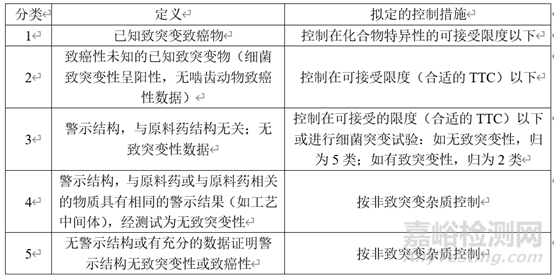
在有關(guān)物質(zhì)中還有一類特殊的雜質(zhì)——DNA反應(yīng)性雜質(zhì)。這一類雜質(zhì)即使在較低水平也能引起DNA損傷�。對(duì)于這一類雜質(zhì)�����,應(yīng)嚴(yán)格控制其含量。依據(jù)ICH M7致癌性及致突變雜質(zhì)可以分為以下5類��。
表3 根據(jù)致突變潛力和致癌潛力對(duì)雜質(zhì)進(jìn)行分類及控制措施
在對(duì)雜質(zhì)進(jìn)行分析時(shí)��,首先檢索文獻(xiàn)和數(shù)據(jù)庫(kù)注����,如果有充分?jǐn)?shù)據(jù)證明警示結(jié)構(gòu)無(wú)致突變性或致癌性則歸為5類,有致突變性�����,但致癌性未知的雜質(zhì)歸為2類��,同時(shí)具有致突變和致癌性的雜質(zhì)歸為1類�����,當(dāng)沒(méi)有致突變的相關(guān)數(shù)據(jù)時(shí)��,則應(yīng)開展細(xì)菌突變?cè)囼?yàn)試驗(yàn)或動(dòng)物試驗(yàn)來(lái)對(duì)雜質(zhì)的致突變性進(jìn)行預(yù)測(cè)或檢測(cè)���,將雜質(zhì)歸為3類��、4類或5類�。
注:如果有Q-SAR預(yù)測(cè)軟件(基于統(tǒng)計(jì)學(xué)和專家規(guī)則各一個(gè)),可以用軟件預(yù)測(cè)替代文獻(xiàn)或數(shù)據(jù)庫(kù)檢索以及細(xì)菌或動(dòng)物試驗(yàn)��,這樣可以提高工作效率�、降低成本。
當(dāng)雜質(zhì)被評(píng)估具有致突變或致癌性時(shí)����,限度要怎樣確定呢?有以下四種計(jì)算方法:
1)基于TTC的可接受攝入量
一個(gè)致突變雜質(zhì)每天攝入1.5 μg所引起的風(fēng)險(xiǎn)被認(rèn)為是可以忽略的����,這一數(shù)值可以適用于大部分藥物。
2)基于雜質(zhì)特異性風(fēng)險(xiǎn)評(píng)估計(jì)算可接受攝入量
①具有陽(yáng)性致癌性數(shù)據(jù)的致突變雜質(zhì)
----可以采用線性外推法:通常以嚙齒類動(dòng)物的TD50除以50000作為雜質(zhì)的可接受攝入量�����,相應(yīng)的患癌風(fēng)險(xiǎn)為1/100000��。
----采用結(jié)構(gòu)相似雜質(zhì)的可接受限度:如果沒(méi)有查到化合物的致癌性數(shù)據(jù)���,但是可以查到與待研究雜質(zhì)化學(xué)結(jié)構(gòu)類似的已知致癌物的致癌性數(shù)據(jù)���,可以采用化合物特異性的方法來(lái)計(jì)算待研究化合物的可接受攝入量����。
②有實(shí)際閾值的致突變雜質(zhì)
可以通過(guò)NOAEL值和不確定因子計(jì)算PDE��,具體計(jì)算方法可以參見ICH Q3C����。
3)與短于終生(LTL)暴露相關(guān)的可接受攝入量
假設(shè)致癌性風(fēng)險(xiǎn)隨暴露量(給藥量)增加而增加��,那么終生以低劑量持續(xù)給藥的患癌風(fēng)險(xiǎn)與累計(jì)暴露量相同的短時(shí)間給藥的患癌風(fēng)險(xiǎn)等同����。因此,短時(shí)間給藥的情況下TTC的值可以高于1.5 μg/天����,這種LTL的概念可適用于臨床研發(fā)階段(要求臨床研究時(shí)間不低于1月)以及上市后有明確預(yù)期治療期限的藥物。
4)多個(gè)致突變雜質(zhì)的可接受攝入量
當(dāng)藥品中含有2個(gè)2類或3類雜質(zhì)���,應(yīng)制定各自的限度�;當(dāng)含有大于等于3個(gè)2類或3類雜質(zhì)�����,應(yīng)總致突變雜質(zhì)限度。限度設(shè)置可參考下表設(shè)置��。
表4 多個(gè)致突變雜質(zhì)可接受攝入量
DNA反應(yīng)性雜質(zhì)的控制方法有4種:
方法1:終點(diǎn)控制�����,即將雜質(zhì)列入終產(chǎn)品質(zhì)量標(biāo)準(zhǔn)���,如果連續(xù)3批生產(chǎn)批或連續(xù)6批中試批樣品中致突變雜質(zhì)水平低于限度30%�,則可以采用定期確認(rèn)行檢測(cè)的方式控制��,否則應(yīng)將該雜質(zhì)的檢測(cè)作為常規(guī)檢測(cè)項(xiàng)��。
方法2:在起始物料或中間體設(shè)置中間控制點(diǎn)����,將雜質(zhì)水平控制在可接受限度內(nèi)。
方法3:在起始物料或中間體設(shè)置中間控制點(diǎn)�����,雜質(zhì)控制標(biāo)準(zhǔn)可高于可接受限度���,結(jié)合工藝控制和對(duì)雜質(zhì)清除和去向的理解確保終產(chǎn)品雜質(zhì)的水平低于可接受限度��。
方法4:基于對(duì)工藝和雜質(zhì)特性的理解����,經(jīng)過(guò)科學(xué)論述,確信終產(chǎn)品中雜質(zhì)水平低于可接受限度����,則無(wú)需對(duì)該雜質(zhì)進(jìn)行檢測(cè)�。
4、小結(jié)
雜質(zhì)研究是貫穿于新藥研發(fā)始終的重要內(nèi)容���,是藥品安全性的重要保障�,近年來(lái)提倡的雜質(zhì)譜研究更是將雜質(zhì)研究上升為科學(xué)系統(tǒng)的研究工程���,在創(chuàng)新藥的研發(fā)中應(yīng)正確認(rèn)識(shí)雜質(zhì)譜研究的重要性�����,在對(duì)雜質(zhì)進(jìn)行全面分析的基礎(chǔ)上結(jié)合安全性評(píng)價(jià)結(jié)果�、穩(wěn)定性特征�����、規(guī)模放大時(shí)雜質(zhì)譜的變化,進(jìn)行風(fēng)險(xiǎn)評(píng)估���,制定科學(xué)合理的控制策略�����,為臨床研究及上市后藥品的安全性提供保障��。
參考文獻(xiàn)
[1]ICH指導(dǎo)原則Q3A��、Q3B��、Q3C�����、Q3D�����、Q6A�����、Q11等����。
[2] ICH 指導(dǎo)原則M7
[3]化學(xué)藥物研究技術(shù)指導(dǎo)原則
[4]仿制藥雜質(zhì)研究與控制的基本思路與策略 張哲峰
[5]淺談雜質(zhì)限度制定的一般原則 張哲峰