[摘要]多肽藥物是一類重要而特別的藥物���。但相比普通小分子化學(xué)藥,目前各國監(jiān)管機構(gòu)和國際組織對多肽化學(xué)藥物的技術(shù)要求尚不完善���。本文就多肽化學(xué)仿制藥質(zhì)量研究的一些特殊技術(shù)要求進行探討,通過一些案例分析提示應(yīng)采用不同原理的分析方法并結(jié)合品種特點全面說明仿制藥與參比制劑的一致性及雜質(zhì)控制策略的合理性�,以提高多肽化學(xué)仿制藥的研究水平和通過率�。
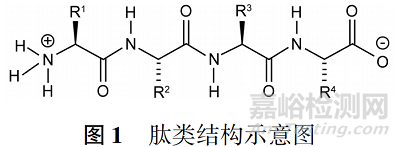
多肽藥物為氨基酸通過肽鍵連接而成的一類化合物(見圖 1) ����,目前尚無法準(zhǔn)確定義�,通常具有明顯的生物活性的多肽都可視為多肽藥物�。多肽藥物可通過從天然物質(zhì)中提取、化學(xué)合成���、天然微生物發(fā)酵及生物重組表達等手段制備得到。從藥品注冊的角度講���,各國監(jiān)管部門均按照多肽的制備方法對這類藥品進行分類�,如化學(xué)合成肽均按照化學(xué)藥品進行管理�,重組表達肽均按照生物制品進行管理等���。此外�,美國 FDA 進一步細分不超過 40 個氨基酸組成的多肽及 41 ~ 99 個全合成多肽為多肽化學(xué)藥物�,41 ~ 99 個非化學(xué)合成多肽或100個以上氨基酸組成的多肽為多肽類生物制品[1]�,其他監(jiān)管機構(gòu)尚未見相關(guān)表述���。目前全球已批準(zhǔn)上市多肽的藥物有 100 種以上,并有多種藥品已在國內(nèi)上市[2]�,涉及生殖、消化�、血液、婦科���、外科����、神經(jīng)�、腫瘤、內(nèi)分泌等各適應(yīng)證領(lǐng)域����,且不乏利拉魯肽����、格拉替雷、戈舍瑞林這樣的重磅品種����,說明國內(nèi)外醫(yī)藥企業(yè)對多肽藥物的研發(fā)和申報充滿熱情����。但與普通小分子藥物相比�,多肽藥物具有分子結(jié)構(gòu)復(fù)雜����、活性高、特異性強的特點���,同時穩(wěn)定性相對較差����,并可能存在諸如免疫原性等的特殊安全性問題���。 這種情況下,對該類藥物的研發(fā)及監(jiān)管提出了更高挑戰(zhàn)����。 本文參考現(xiàn)有技術(shù)法規(guī)����,特別是國際監(jiān)管同行的研究及審評經(jīng)驗�,結(jié)合相關(guān)文獻案例,對一些多肽化學(xué)仿制藥質(zhì)量研究過程中需要關(guān)注的一些特殊技術(shù)要求進行簡介�,希望為我國多肽化學(xué)仿制藥的研發(fā)和監(jiān)管提供一些有意義的借鑒���。
一�、多肽化學(xué)仿制藥的監(jiān)管現(xiàn)狀
國相比普通小分子化學(xué)藥���,目前各國監(jiān)管機構(gòu)和規(guī)際組織對多肽化學(xué)藥物的技術(shù)要求尚不完善,法舉文件相對缺乏�,存在一定的監(jiān)管空白�。 表 1 中列了各國及國際監(jiān)管機構(gòu)目前僅有的幾個涉及多肽化學(xué)藥物的官方技術(shù)支持文件���。 其中國家藥品監(jiān)督管理局(以下簡稱國家局) 早在 2007 年就出臺了《合成多肽藥物藥學(xué)研究技術(shù)指導(dǎo)原則》�,說明我國很早就開始重視這類藥物的研發(fā)和監(jiān)管問題����,同時在《化學(xué)藥物原料藥制備和結(jié)構(gòu)確證研究技術(shù)指導(dǎo)原則》中也提到了關(guān)于多肽藥物的一些關(guān)鍵技術(shù)要求����。 上述指導(dǎo)原則在出臺后的 10 多年間對我國多肽化學(xué)藥物的發(fā)展起到了一定的促進作用�, 但由于種種原因未能及時升級更新,已不能充分滿足目前多肽藥物研究與審評的需要����,且在我國仿制藥一致性評價的大背景下,考慮到多肽藥物的復(fù)雜性����,上述指導(dǎo)原則在評價仿制藥與參比制劑的一致性方面還有進一步完善的空間�。 最近,國家藥品監(jiān)督管理局藥品審評中心為進一步指導(dǎo)化學(xué)合成多肽藥物的藥學(xué)研究�,提供可參考的技術(shù)標(biāo)準(zhǔn)����,于 2023 年 2 月發(fā)布了《化學(xué)合成多肽藥物藥學(xué)研究技術(shù)指導(dǎo)原則(試行)》[3]�,為未來一段時間內(nèi)我國多肽仿制藥的研發(fā)和監(jiān)管提供了依據(jù)。 國外監(jiān)管機構(gòu)中�,ICH Q系列指導(dǎo)原則中的 Q3A 和 Q3B 明確不適用于肽類藥物����;Q6A 顯示適用于小于 10 個氨基酸的低分子量合成肽,但不適用于高分子肽和多肽����,且并未就多肽化學(xué)藥物的特殊要求進行專門闡述;Q6B 明確不適用于合成肽���。 歐盟未曾發(fā)布專門針對多肽化學(xué)藥物的指導(dǎo)原則����,僅在抗生素藥物雜質(zhì)限度指導(dǎo)原則及歐洲藥典藥用物質(zhì)通則中提到了多肽藥物雜質(zhì)控制的一般要求。 美國 FDA 曾于 1994 年頒布過關(guān)于遞交合成多肽原料藥申請藥學(xué)資料的相關(guān)要求�,但目前該文件已失效。 不過美國 FDA 近期就多肽化學(xué)仿制藥的研究公布了多個復(fù)雜多肽仿制藥的個藥指南���,并在 2021 年頒布了《仿制重組 DNA 來源藥物的高純度合成多肽藥物指導(dǎo)原則》[4],說明國際監(jiān)管機構(gòu)也開始逐漸致力于完善多肽仿制藥監(jiān)管體系的建設(shè)工作�。

二、多肽化學(xué)仿制藥質(zhì)量研究的特殊技術(shù)要求
多肽化學(xué)仿制藥的藥學(xué)研究首先應(yīng)遵循國家局已經(jīng)發(fā)布的關(guān)于化學(xué)仿制藥一致性評價相關(guān)技術(shù)指南的一般要求���,在此不過多贅述����。 但由于多肽主要由氨基酸構(gòu)成�,其中化學(xué)合成肽還會引入很多非天然氨基酸,這使得這類藥物在制備方法���、結(jié)構(gòu)確證����、質(zhì)量研究等方面均存在與一般化學(xué)小分子藥物不同的問題。
首先多肽為氨基酸組成的鏈狀���、分支狀或環(huán)狀結(jié)構(gòu)�,因此結(jié)構(gòu)確證時除運用元素分析����、光譜、質(zhì)譜等常規(guī)方法外���,需要結(jié)合結(jié)構(gòu)特點采用一些不同于小分子化學(xué)藥的分析手段����,包括但不限于:采用氨基酸組成和手性分析����、序列分析或肽圖測繪等方法確定一級結(jié)構(gòu),采用圓二色散���、熒光光譜����、光散射����、X?射線衍射等確定高級結(jié)構(gòu)等���,仿制藥除一級結(jié)構(gòu)氨基酸序列必須與參比制劑相同外,高級結(jié)構(gòu)也需要與后者保持一致����。 另外,需要關(guān)注的是有些多肽藥物存在超分子聚集為寡聚物或多聚物的現(xiàn)象����,該性質(zhì)與多肽藥物的活性及體內(nèi)藥動學(xué)行為均有較大關(guān)系���,如何表征仿制藥與參比制劑聚集特性和聚集動力學(xué)的一致存在一定難度���,現(xiàn)有的研究方法包括分子排阻色譜法、光散射法���、熒光分析法���、分析超速離心法����、凝膠電泳法����、場流分離法����、核磁共振法等���。 同時,也應(yīng)關(guān)注仿制藥與參比制劑在一些特殊物化特性(如分子量分布�、表面電荷分布、黏度���、光密度等)上的一致性���。
在確保仿制藥與參比制劑結(jié)構(gòu)或物質(zhì)基礎(chǔ)等同的前提下�,二者的降解雜質(zhì)譜應(yīng)無較大差異����,但由于仿制藥的制備工藝往往不可能完全等同于參比制劑����,導(dǎo)致工藝雜質(zhì)可能明顯不同,特別是一些結(jié)構(gòu)相似的肽類雜質(zhì)����,這些雜質(zhì)的定性與定量研究成為多肽化學(xué)仿制藥質(zhì)量能否合理控制的關(guān)鍵,同時考慮到肽類雜質(zhì)可能存在與一般小分子化藥不具有的異常毒性和免疫原性���,因此雜質(zhì)限度的確定更應(yīng)有充分依據(jù)���。 此外聚合物雜質(zhì)為多肽藥物中普遍存在的一種雜質(zhì),在工藝及貯藏過程中均可能產(chǎn)生���,考慮到這種雜質(zhì)更易引起特殊安全性,因此對仿制藥與參比制劑聚合物雜質(zhì)的對比研究應(yīng)加以重視�。
三、多肽化學(xué)仿制藥質(zhì)量研究的案例分析
3.1硫酸魚精蛋白注射液
該品種于20 世紀(jì) 80年代之前已在美國上市�,用于因注射肝素過量引起的出血。 目前國內(nèi)有 3 家仿制藥上市����,但獲批時間均較早�,且尚未進行一致性評價。 其原料藥為從大馬哈魚體內(nèi)提取的一種堿性蛋白質(zhì)的硫酸鹽����,為多肽混合物,其中 4 種主要多肽(見表 2)總含量超過90% �,剩余由次要多肽組成����。 由于含有多個成串的精氨酸���,且 4 種主要多肽氨基酸序列非常相似�,因此分離純化和質(zhì)量分析均存在難度�。 考慮到魚源及提取工藝的不同可能會造成仿制藥的原料藥多肽組成與參比制劑的差異�,因此應(yīng)采用適當(dāng)?shù)姆椒ㄈ娑ㄐ约岸糠治霎a(chǎn)品組成并與參比制劑對比�,以確保仿制藥質(zhì)量符合要求����。
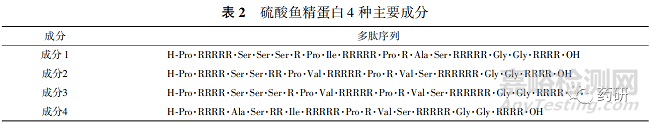
原研公司曾報道[4]�,使用 pH = 2 的磷酸鹽緩沖液和低含量的乙腈作為洗脫劑對原料藥各組分進行液相分離再進行序列分析的方法�,可確定本品中 4個主要多肽的氨基酸組成和比例,并推測是二級結(jié)構(gòu)中轉(zhuǎn)角處脯氨酸周圍殘基的差異導(dǎo)致了結(jié)構(gòu)相似組分間色譜行為的不同�,同時提示該方法也有望用于本品的大規(guī)模生產(chǎn)���。 除色譜方法外����,美國 FDA 認為仿制藥的研究還需要有更多的考量。 他們曾以 5種不同廠家的硫酸魚精蛋白為對象進行研究[6]�,初步結(jié)果表明�,不同廠家原料藥表現(xiàn)出一定的熱力學(xué)性質(zhì)(如失水量、玻璃轉(zhuǎn)化溫度����、熔點等)的差異�,但譜學(xué)性質(zhì)(如紅外���、圓二色譜���、一維核磁等) 差異不明顯�,均顯示出該原料藥在溶液中呈現(xiàn)一種無規(guī)則卷曲加少量 β 片層的構(gòu)象形態(tài),同時基于 QbD 原則所建立的色譜方法也可有效表征不同廠家原料藥 4種主要多肽組分及含量上與參比制劑的一致性����。 但隨后美國 FDA 研究人員重新審視了研究方法的合理性���,進一步認為由于該原料藥缺乏明顯的二級結(jié)構(gòu)�,紅外和圓二色譜方法可能對序列的微小變化較不敏感�,因此利用核磁共振譜和質(zhì)譜作為與色譜方法的正交分析手段重新進行了評估,以提供進一步的支持[7]���。 通過測定1HNMR 中 Arg���,Ala 和 Gly 等關(guān)鍵氨基酸相應(yīng)特征信號的積分比值,并分析質(zhì)譜中主成分特征峰的質(zhì)荷比和相對含量����,再次確認了前次研究的結(jié)論�。 此外,采用核磁同核或異核二維相關(guān)譜還可建立用于本品質(zhì)控的指紋圖譜���。 上述方法的綜合運用,對有效鑒別不同來源硫酸魚精蛋白間的定性及定量差異有一定的參考意義���,而且還可有效區(qū)分正品與假劣產(chǎn)品(如鯡魚來源的魚精蛋白或哺乳動物來源的蛋白等)。
3.2 醋酸地加瑞克
該品種分別于 2008 和 2009年在美國和歐洲上市���,于 2018 年批準(zhǔn)進口我國���,為第 3 代促性腺激素釋放激素(GnRH) 受體阻斷劑�,對雄激素依賴性腫瘤有治療作用����,適用于需要雄激素去勢治療的前列腺癌患者,目前國內(nèi)外均無仿制藥上市�。 其原料藥為化學(xué)合成線性十肽(圖 2),含7 個非天然氨基酸����。原料藥易溶于水,但室溫下在0. 1 ~ 10 mg·mL-1濃度的水溶液中會自發(fā)聚集�,4h后呈凝膠狀。 該過程受濃度�、時間、溫度���、鹽含量和蛋白質(zhì)等因素的影響�,因此其制劑凍干粉復(fù)溶后雖在體外 1 h 內(nèi)穩(wěn)定性良好,但皮下給藥接觸體液后立即形成凝膠�,藥物從凝膠中緩慢釋放,從而起到長效給藥的作用[8]����。目前發(fā)現(xiàn)GnRH類似物在水溶液中均有自聚集現(xiàn)象����,聚集體的結(jié)構(gòu)和形成凝膠的性質(zhì)因化合物而異。 地加瑞克由于為兩性分子����,主要是通過親水端和疏水端相互結(jié)合,并延長聚集形成纖維網(wǎng)絡(luò)最終形成凝膠���。 原料藥的這種有條件聚集的特性對制劑最終的安全有效性影響極大����,藥學(xué)研究與評價均存在一定的難度�。
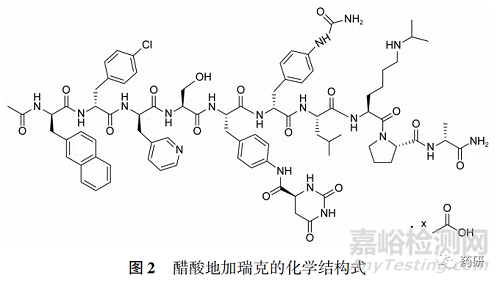
原研公司對地加瑞克的聚集狀態(tài)進行了結(jié)構(gòu)表征[8],結(jié)果表明其聚合物呈纖維狀���,纖維直徑為5~15 nm���,且典型的 X?射線衍射圖中呈現(xiàn) 4. 6 ~ 4. 8 Å的子午線方向衍射���,提示了在縱向上的有序性�。 紅外圖譜顯示�,無論在體內(nèi)還是體外,所形成的淀粉樣聚合纖維均顯示出明顯的 β 折疊結(jié)構(gòu)����,但最大吸收峰波數(shù)略有不同,提示體內(nèi)外纖維的高級結(jié)構(gòu)在微觀上略有差異�。 Maji 等[9] 通過生化染色(如硫磺素T 染色����、剛果紅染色等)及電鏡觀察,進一步證實了地加瑞克在體外的聚集性質(zhì),并通過透析試驗定量證明了本品的釋藥方式(即從形成的淀粉樣纖維末端逐漸釋放藥物單體)�。 原研品歐盟審評報告[10]及原研公司公開專利[11]均顯示,基于原料藥的此種聚集特性�,結(jié)合統(tǒng)計分析說明制劑體外加速釋放度試驗與光密度和黏度的實測值間具有相關(guān)性,原研公司在制劑標(biāo)準(zhǔn)中僅通過對成品部分物化性質(zhì)(如復(fù)溶溶液的黏度和光密度等) 進行相應(yīng)控制�,就可確保原研品在體內(nèi)釋放行為的批間一致���。
但仿制藥證明自身與參比制劑的一致僅此還不夠充分�。 美國FDA于2021 年3 月提出了該品種的個藥指南草案稿[12]�,明確本品的仿制藥首先需滿足與參比制劑 Q1 和 Q2 的要求(即注射劑中輔料種類和用量應(yīng)與參比制劑相同),其次需證明原料藥一級和二級結(jié)構(gòu)及聚集性質(zhì)與參比制劑的一致性�,并需要對制劑的必要物化性質(zhì)(如復(fù)溶時間、乙酸含量���、外觀、光密度����、黏度���、pH 等)�、聚集動力學(xué)和體外釋放行為進行全面對比研究���,其中聚集動力學(xué)應(yīng)在適當(dāng)?shù)臈l件如緩沖溶液或模擬體內(nèi)介質(zhì)中進行����,體外藥物釋放試驗應(yīng)說明試驗設(shè)計的合理性����,并提供必要的方法學(xué)研究和驗證資料�。各項要求中����,聚集特性和聚集動力學(xué)為研究的重點及難點�。除可采用上文已提及的生化方法進行評價外����,美國 FDA 研究人員還建立了一種基于核磁共振的試驗方法[13]����。該方法提示����,實時1HNMR中高場區(qū)0~2 ppm間特征烷基信號的展寬可定性說明溶液中聚集結(jié)構(gòu)正逐漸形成����,且峰面積的變化與地加瑞克早期納米尺度下體外聚集的速率間存在數(shù)量關(guān)系,該方法對地加瑞克的一些體外聚集特征(如濃度、pH 值����、鹽和溫度對聚集的影響)均可進行合理解釋,因此有望用于評價仿制藥與參比制劑的一致性�。 隨著技術(shù)的進步,相信仍會有更多更好的研究方法不斷涌現(xiàn)����。
3.3 醋酸格拉替雷
該品種于 1996 和 2001 年分別在美國和歐洲上市,且已有多個仿制藥獲批�,用于治療多發(fā)性硬化癥,但在我國原研及仿制藥均未上市���。 其原料藥為由谷氨酸����、賴氨酸�、丙氨酸和酪氨酸以一定比例(約 0. 14∶ 0. 34∶ 0. 43∶ 0. 09)組成的肽段共聚 混 合 物, 其 結(jié) 構(gòu) 為: ( Glu�, Ala, Lys���, Tyr )x·xCH3COOH���,平均分子量為 5 ~ 9 kDa����。 文獻[14]顯示�,其合成步驟首先為單個氨基酸活化并與引發(fā)劑結(jié)合,其次在一定條件下 4 種氨基酸以近乎隨機方式發(fā)生聚合���,形成多肽混合物�,再在一定條件下發(fā)生部分解聚�,考慮到各氨基酸反應(yīng)活性存在差異,因此反應(yīng)條件的選擇對最終產(chǎn)品的物質(zhì)組成有較大影響�,原研原料藥在批與批之間也存在一定范圍內(nèi)的差異,仿制藥與參比制劑質(zhì)量一致性比較上如何要求也具有相當(dāng)?shù)奶魬?zhàn)����。
對于本品這樣的復(fù)雜藥物,采用多肽藥物質(zhì)量研究的一般方法不足以比較仿制藥和參比制劑的質(zhì)量����。 為此����,美國 FDA 專門發(fā)布了個藥指南[15]�,明確在確保仿制藥組成及理化性質(zhì)與參比制劑一致的基礎(chǔ)上����,還需要結(jié)合制備工藝、起始原料和過程控制及生物活性測試結(jié)果進行綜合評價����。 首先,仿制藥的制備工藝應(yīng)與參比制劑一致或相當(dāng)����,特別強調(diào)了起始原料氨基酸活化形式和聚合反應(yīng)引發(fā)劑及酸裂解反應(yīng)試劑應(yīng)等同;其次���,產(chǎn)品物化性質(zhì)需一致���,包括4 種氨基酸的含量和光學(xué)純度、分子量分布���、指紋圖譜特征(如紅外���、圓二色散、核磁共振等)�;再次���,聚合和解聚的反應(yīng)特征應(yīng)相當(dāng),應(yīng)對聚合開始����、聚合過程中鏈增長、部分解聚裂解過程中的物料結(jié)構(gòu)進行詳細研究并與參比制劑具有可比性����;最后,典型體外動物模型上表現(xiàn)出的生物活性應(yīng)等效����。 不過值得注意的是,考慮到參比制劑本身的復(fù)雜性����,在與其進行充分對比研究的基礎(chǔ)上,該指南允許仿制藥的相關(guān)特性在一定范圍內(nèi)發(fā)生變動���。
有部分文獻探討了如何在該指南的基礎(chǔ)上去評價仿制藥與參比制劑的一致性�。 其中���,對于物質(zhì)基礎(chǔ)的等同�,美國 FDA 的研究人員提供了一種解決方案[16]���。 采用包括核磁共振����、非對稱性場流多角度光散射分離檢測和液質(zhì)聯(lián)用等不同分析方法的組合�,對比研究 3 批參比制劑及 1 批各氨基酸比例(酪氨酸和賴氨酸比例與參比制劑有 2% 的出入)與分子量(遠高于參比制劑)均與參比制劑不同的工具化合物。 結(jié)果顯示所用方法可有效檢測出 2 種藥物的分子量及氨基酸比例及序列的差異����,且具有統(tǒng)計學(xué)意義。 該研究提示�,若干高靈敏度的正交分析方法的組合運用,可能是分析這類極其復(fù)雜的原料藥的一種解決之道����。
此外,從 Mylan 公司在歐盟獲批上市的本品仿制藥審評報告[17] 也可以看出歐盟對該品種的審評思路����。 仿制藥藥企首先通過嚴(yán)格控制合成工藝參數(shù)及合理擬定放行標(biāo)準(zhǔn)確保了仿制藥批間的一致性;其次采用經(jīng)驗證的方法說明仿制藥與參比制劑一級及高級結(jié)構(gòu)的相似�,并證明了該方法對具有相似組成但不同工藝制備樣品的區(qū)分力;再次分別對仿制藥與參比制劑中的 9 個主要成分進行分離�,通過多種化學(xué)和生物學(xué)手段構(gòu)建了用于評價一致性或相似性的指紋圖譜����,最后借助成品及各主要成分體內(nèi)外生物學(xué)研究及臨床研究進一步說明了仿制藥與參比的相似性����。 體外藥學(xué)部分整體評價思路與美國FDA 基本一致,但歐盟認為如此復(fù)雜仿制藥的批準(zhǔn)可能仍需要臨床試驗數(shù)據(jù)的支持�。
原研公司采用 7 種物理化學(xué)方法及 7 種生物學(xué)方法綜合評價了美國和歐洲上市仿制藥與參比制劑的相似性[18 - 19]。 采用低靈敏度的理化方法(如分子量分布�、考馬斯亮藍染色等) 檢測結(jié)果顯示二者之間無明顯差異,但采用高靈敏度的方法(如反相色譜多角度光散射法���、陽離子交換色譜法等) 檢測結(jié)果顯示仿制藥組成和比例均與參比制劑略有不同����,此外�,仿制藥細胞體外試驗活性要略高于參比制劑,而動物體內(nèi)研究則顯示仿制藥在給藥部位局部毒性的出現(xiàn)頻率也高于參比制劑����。 原研公司的研究結(jié)果進一步印證了這類品種的極端復(fù)雜性,無論研發(fā)或是監(jiān)管均需特別審慎���。
3.4鮭降鈣素
該品種于20 世紀(jì) 80 年代前在美國上市�,用于治療骨質(zhì)疏松。 其原料藥為來源于大馬哈魚體內(nèi)的一種 32 肽���,結(jié)構(gòu)中含 1 個二硫鍵(圖3),目前上市品來源多為化學(xué)合成或生物重組���。 目前已有多個仿制藥在我國上市���,但上市時間均較早,且尚未進行一致性評價�。 該品種結(jié)構(gòu)或物化特性相對并不復(fù)雜,但由于仿制藥制備工藝與原研藥明顯不同�,因此更需關(guān)注雜質(zhì)控制的合理性,特別是來源于制備工藝的特有雜質(zhì)����。 對于多肽化學(xué)藥物雜質(zhì)研究來說,通常需同時綜合應(yīng)用不同原理的方法進行交叉研究���,重點關(guān)注方法對肽類雜質(zhì)的分離和檢出能力�。

美國藥典(USP)鮭降鈣素原料藥各論推薦采用高效液相色譜法控制合成來源鮭降鈣素原料藥中的有關(guān)物質(zhì)���,但未規(guī)定特定雜質(zhì)����,僅控制一般單雜與總雜;制劑各論中未對有關(guān)物質(zhì)進行控制�。 美國 FDA結(jié)合審評經(jīng)驗,建立液質(zhì)聯(lián)用方法對已上市某種鮭降鈣素鼻腔噴霧劑進行研究[20]���,檢測到該產(chǎn)品中總雜水平要遠高于 USP 推薦的有關(guān)物質(zhì)方法的檢出水平����,同時新方法還可對各已知雜質(zhì)及 10 個未知雜質(zhì)進行定性和定量����,表明新方法較藥典方法具有更高的雜質(zhì)分離和檢出能力,也印證了采用不同原理的方法進行多肽藥物雜質(zhì)研究工作的必要性�。 美國FDA 利用該方法對已上市的 13 種鮭降鈣素鼻噴劑進行檢驗,結(jié)果表明不同來源制劑的質(zhì)量參差不齊���,為進一步加強監(jiān)管及提升行業(yè)總體研究水平提供了依據(jù)����。 此外����,Windisch 等[21]報道鮭降鈣素在高溫條件下的降解實驗中有明顯的二聚物或多聚物雜質(zhì)生成�,這類聚合物雜質(zhì)在工藝過程中�,特別是當(dāng)仿制藥原料藥制備工藝或制劑所用輔料與參比制劑有較大差異時,都有可能產(chǎn)生����。 這類雜質(zhì)對多肽藥物的安全性有較大影響�,因此需要特別關(guān)注。
從上面幾個例子可以看到���,對于復(fù)雜多肽藥物���,特別是涉及多組分、存在高級結(jié)構(gòu)或具有聚集特性的多肽���,在進行仿制藥研發(fā)時����,應(yīng)采用不同原理的分析方法并結(jié)合品種特點對比參比制劑進行研究���,以全方位說明仿制藥與參比制劑的一致性����,必要時還應(yīng)選擇合適的指標(biāo)在質(zhì)量標(biāo)準(zhǔn)中進行相應(yīng)控制。 另外���,在仿制藥研發(fā)早期即應(yīng)對工藝可能引入的雜質(zhì)包括聚合物雜質(zhì)采用適宜方法進行充分研究����,根據(jù)研究結(jié)果制定合理的控制策略���,雜質(zhì)的限度應(yīng)以參比制劑雜質(zhì)實際水平為依據(jù)����,并參照相關(guān)指南合理擬定����,必要時關(guān)注雜質(zhì)的特殊安全性。 還應(yīng)注意的是���,有些原研多肽由于上市較早����,當(dāng)時研究手段有限����,質(zhì)量研究和控制標(biāo)準(zhǔn)放在當(dāng)下可能并不充分���,在進行仿制藥開發(fā)時還應(yīng)基于產(chǎn)品的結(jié)構(gòu)特點以及結(jié)構(gòu)與療效、安全性的關(guān)系�,建立新的質(zhì)量研究方法、調(diào)整控制策略�。
四、結(jié)語
綜上所述����,多肽仿制藥的研究應(yīng)符合仿制藥研究的一般要求����,同時要考慮其技術(shù)上的特殊性,仿制藥結(jié)構(gòu)和物化特性與參比制劑的等同及對雜質(zhì)的合理控制是獲得成功的關(guān)鍵�,應(yīng)結(jié)合品種特點科學(xué)制定研究方案。 目前市場上多肽藥品數(shù)量很多���,但質(zhì)量良莠不齊����,多肽藥物的研發(fā)和監(jiān)管水平均亟須提升���。 2018 年國務(wù)院辦公廳發(fā)布了《關(guān)于改革完善仿制藥供應(yīng)保障及使用政策的意見》[22]���,提出要制定鼓勵仿制的藥品目錄����,目前已發(fā)布的2批目錄[23 - 24]共計50個品種中�,就包括了格拉替雷和普卡那肽等多個多肽藥物品種,說明國家非常重視多肽仿制藥的發(fā)展���。同時該文件還要求健全產(chǎn)學(xué)研醫(yī)用協(xié)同創(chuàng)新機制�,建立仿制藥技術(shù)攻關(guān)聯(lián)盟�,發(fā)揮企業(yè)的主導(dǎo)作用和醫(yī)院、科研機構(gòu)�、高等院校的基礎(chǔ)支撐作用,加強仿制藥的技術(shù)攻關(guān)����。 希望通過本文與業(yè)界對多肽化學(xué)仿制藥進行交流,并促進更多更好的多肽仿制藥加快上市����,提高我國患者的用藥可及性。