臨床試驗數據質量是評價新藥有效性和安全性的關鍵���,完整數據鏈貫穿臨床試驗全過程。數據溯源是對數據的追本溯源�,以實現對歷史數據的重現�。
2023年5月17日,廣東省藥學會公眾號廣東藥物臨床試驗發(fā)布《藥物臨床試驗 源數據管理·廣東共識(2023版征求意見稿)》���,供各醫(yī)療機構和臨床試驗相關方參考�。新版《藥物臨床試驗質量管理規(guī)范》(2020版)(2020年第57號)自2020年7月1日起施行���,對臨床試驗的管理帶來新挑戰(zhàn)和新問題�,該指南對臨床試驗中涉及源數據/源文件的管理要求提出了一些業(yè)內認可的做法和建議�,明確源數據、源文件的概念�、類別以及載體形式�,統(tǒng)一申辦方/CRO、研究者、試驗機構管理人員等各方對源數據管理的認識差異�,提高臨床試驗數據管理的質量���。
1、源文件/源數據/元數據你能分清嗎���?
在藥物臨床試驗的日常工作中,我們幾乎每天都在和臨床試驗中的文件與數據打交道�,了解清楚源文件/源數據/元數據的概念可以使工作更加得心應手����。
1.1源文件(Source Documents)
GCP定義:指臨床試驗中產生的原始記錄�、文件和數據���,如醫(yī)院病歷、醫(yī)學圖像�、實驗室記錄���、備忘錄�、受試者日記或者評估表、發(fā)藥記錄���、儀器自動記錄的數據����、縮微膠片���、照相底片����、磁介質�、X光片、受試者文件���,藥房�、實驗室和醫(yī)技部門保存的臨床試驗相關的文件和記錄���,包括核證副本等。源文件包括源數據����,可以以紙質或者電子等形式的載體存在����。(來源:2020年7月1日實施的《藥物臨床試驗質量管理規(guī)范》(2020年第57號))����。
廣東共識版定義:是指包括了源數據的文件,可以是原始文件或其核證副本,以紙質或者電子等形式存在���。比如醫(yī)院病歷�、醫(yī)學圖像�、實驗室記錄���、備忘錄、受試者日記或者評估表����、發(fā)藥記錄����、儀器自動記錄的數據����、縮微膠片、照相底片����、磁介質、X光片����、受試者文件,藥房���、實驗室和醫(yī)技部門保存的臨床試驗相關的文件和記錄(CRF中的數據不是源數據,通常不作為源文件)���。(來源:藥物臨床試驗 源數據管理·廣東共識(2023版征求意見稿))
1.2源數據(Source Data)
GCP定義:指臨床試驗中的原始記錄或者其復印件(即核證副本���,指經過審核驗證�,確認與原件的內容和結構等均相同的復制件���,該復制件是經審核人簽署姓名和日期���,或者是由已驗證過的系統(tǒng)直接生成�,可以以紙質或者電子等形式的載體存在�。核證副本也可以作為有效的記錄)上記載的所有信息,包括臨床發(fā)現���、觀測結果以及用于重建和評價臨床試驗所需要的其他相關活動記錄����。
廣東共識版定義:指臨床試驗中的原始記錄或其核證副本上記載的所有信息�,包括臨床發(fā)現���、觀測結果以及用于重建和評價臨床試驗所需要的其他相關活動記錄。源數據應當具有可歸因性���、易讀性�、同時性、原始性����、準確性、完整性����、一致性和持久性。(來源:藥物臨床試驗 源數據管理·廣東共識(2023版征求意見稿))
1.3元數據(Meta Data)
是用來定義和描述數據的數據�,通過定義和描述數據����,可以支持對其所描述的數據對象的定位、查詢���、交換���、追蹤、訪問控制�、評價和保存等諸多管理工作����。(來源:2020年12月1日實施的《藥品記錄與數據管理要求(試行)》)
2、藥物臨床試驗中源數據和源文件管理
在臨床試驗中,源數據是作為溯源依據的數據����,源文件是承載源數據的原始文件����,自開展臨床試驗數據現場核查以來,藥監(jiān)管理部門不斷強調臨床試驗數據的產生�、收集、記錄和報告過程應真實���、完整和準確�。臨床試驗中立項�、倫理、過程中以及結題等�,都離不開源數據管理,而這些源數據管理都符合相關要求嗎����?
2.1源數據的類別和管理要求
2.2源文件的類別和管理要求
2.3源數據如何鑒定?
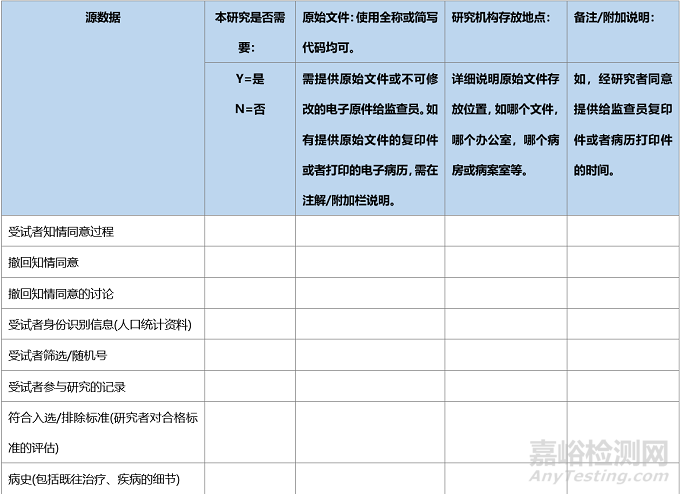
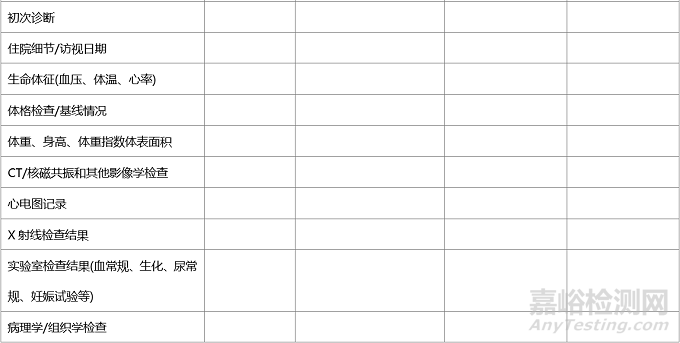
在臨床試驗中,同一事件的數據可能會由不同人獲取并記錄���,如臨床護士和研究醫(yī)生可能都會采集試驗中受試者的生命體征����,并分別記錄在護理單和病歷中�,或由同一人獲得并記錄在不同的地方���,如研究人員將采集的生命體征分別記錄在臨床病歷和受試者文件中����,為確證病例報告表(CRF)所記錄數據的有效來源,建議在臨床試驗開始前�,采用“源數據鑒認表”���,源數據鑒認表可參見以下范例�。
表.臨床試驗-源數據鑒認表建議范例
本研究整個持續(xù)時間內�,如果原始文件的存放位置發(fā)生變化�,監(jiān)查員�、研究者或指定人員需更新此表���,或需要用新版本表格記錄發(fā)生變化的信息�,要保留所有既往版本,并標記為“被取代”����。
3、臨床試驗核查中源數據管理監(jiān)管關注點
國家藥品監(jiān)督管理局頒布的《藥品注冊核查要點與判定原則(藥物臨床試驗)(試行)》(2022年1月1日實施)是藥品臨床試驗項目數據核查的金標準����。檢查要點包含2個部分11大項:
3.1藥物臨床試驗-臨床試驗部分(8大項)
1) 臨床試驗許可與條件
2) 倫理審查
3) 臨床試驗實施過程
4) 試驗用藥品管理
5) 生物樣品管理
6) 中心實驗室及獨立評估機構
7) 臨床試驗數據采集與管理
8) 委托研究
3.2藥物臨床試驗-生物樣品分析部分(3大項)
9) 生物樣品分析條件與合規(guī)性
10) 生物樣品分析實驗的實施
11) 記錄的管理
《藥品注冊核查要點與判定原則(藥物臨床試驗)(試行)》對研究過程中原始記錄和數據進行核實、實地確認����,經核查確認發(fā)現以下情形之一的,核查認定為“不通過”�,11種情形如下:
編造或者無合理解釋地修改受試者信息以及試驗數據、試驗記錄�、試驗藥物信息����;
以參比制劑替代試驗制劑����、以試驗制劑替代參比制劑或者以市場購買藥品替代自行研制的試驗用藥品,以及以其他方式使用虛假試驗用藥品���;
隱瞞試驗數據���,無合理解釋地棄用試驗數據,以其他方式違反試驗方案選擇性使用試驗數據����;
瞞報可疑且非預期嚴重不良反應;
瞞報試驗方案禁用的合并藥物���;
故意損毀����、隱匿臨床試驗數據或者數據存儲介質����;
關鍵研究活動����、數據無法溯源����;
申報資料與原始記錄不一致且影響結果評價;
其他嚴重數據可靠性問題�;
拒絕、不配合核查����,導致無法繼續(xù)進行現場核查�;
法律法規(guī)規(guī)定的其他不應當通過的情形。
參考資料:
1.www.sinopharmacy.com.cn