1���、生產(chǎn)工藝
在考慮除菌過濾系統(tǒng)工藝時����,應注意除菌過濾工藝的局限性����。除菌過濾器不能將病毒或支原體全部濾除,因此在生產(chǎn)工藝中要考慮彌補除菌過濾的這些不足����。
為了降低過濾除菌的風險���,需要安裝第二只已滅菌的除菌過濾器�。在除菌過濾器完成工藝過濾以后���,這兩只過濾器中至少有一個要通過完整性檢查�。除菌過濾應在規(guī)定的工藝控制參數(shù)范圍內進行,同時為保證除菌過濾的有效性�,還應對影響除菌過濾效果的關鍵參數(shù)進行控制和記錄??刂祈椖繎ǔ^濾溫度、時間����、壓力、上下游壓差等����。最終的除菌過濾器應當盡可能接近灌裝點,即盡可能地省略除菌過濾后料液的中間儲存環(huán)節(jié)�。
使用前,除菌過濾器必須經(jīng)過滅菌處理(如在線或離線蒸汽滅菌�、輻射滅菌等)。在線蒸汽滅菌的設計及操作過程應重點考慮濾芯可耐受的最高壓差及溫度。使用滅菌器進行滅菌時���,通常采用脈動真空滅菌方法。滅菌過程應保證濾器能被蒸汽穿透���,從而達成對過濾器的徹底滅菌�。除菌過濾系統(tǒng)應盡可能地考慮采用在線滅菌�,如果仍需采用離線滅菌后再裝配的話,建議使用無菌連接器來降低無菌風險�。
此外����,尚在產(chǎn)品研發(fā)階段���,就需要對除菌濾芯進行細菌截留試驗�、化學兼容性試驗�、可提取物或浸出物試驗�、安全性評估和吸附評估等研究���。對過濾過程中潛在可能引入的雜質和風險進行充分的評估����。
1.2模擬灌裝
培養(yǎng)基模擬灌裝是非最終滅菌無菌制劑的關鍵驗證點���,不符合規(guī)范要求的無菌工藝過程,不能通過模擬試驗來證實其無菌控制措施的合理性�。
對于凍干制劑���,需要在液體灌裝的基礎上增加冷凍干燥工藝。培養(yǎng)基模擬灌裝試驗應從第一步無菌操作開始����,直至無菌產(chǎn)品完全密封結束�。
在進行基于無菌風險的模擬試驗方案設計時,應注意設計要結合無菌生產(chǎn)工藝���,盡量與實際無菌操作過程保持一致�,以求試驗結果真實反映生產(chǎn)過程的無菌保證水平����。
在無菌生產(chǎn)工藝中,暴露操作對最終產(chǎn)品的無菌特性有著重要影響����。如設備(或管道)的無菌連接�、無菌容器的轉運和更換���、灌裝等關鍵操作等����,都是引入微生物污染的重點環(huán)節(jié)及風險點���。因此,模擬試驗方案設計應對這些過程中無菌防護措施的有效性進行仔細考察�。
為了確認無菌生產(chǎn)工藝中無菌質量風險控制的有效性�,通常采用生產(chǎn)過程中可能發(fā)生的最差條件進行模擬試驗���。值得注意的是,最差條件并不是指人為創(chuàng)造的超出允許范圍的生產(chǎn)狀況和環(huán)境����。最差條件應基于風險等級并結合無菌生產(chǎn)工藝����、設備裝備水平、人員數(shù)量和干預等因素來設計。
1.3清潔驗證
在考慮清潔驗證前,應先確保廠房���、生產(chǎn)設施和設備滿足2010版藥品生產(chǎn)質量管理規(guī)范(GMP)中的基本要求:
• 生產(chǎn)特殊性質的藥品����,如高致敏性藥品(如青霉素類)或生物制品(如卡介苗或其他用活性微生物制備而成的藥品),必須采用專用和獨立的廠房�、生產(chǎn)設施和設備���。
• 青霉素類藥品產(chǎn)塵量大的操作區(qū)域應當保持相對負壓,排至室外的廢氣應當經(jīng)過凈化處理并符合要求���,排放口應當遠離其他空氣凈化系統(tǒng)的進風口。
• 生產(chǎn)β-內酰胺結構類藥品、性激素類避孕藥品必須使用專用設施(如獨立的空氣凈化系統(tǒng))和設備���,并與其他藥品生產(chǎn)區(qū)嚴格分開�。
• 生產(chǎn)某些激素類����、細胞毒性類�、高活性化學藥品應當使用專用設施(如獨立的空氣凈化系統(tǒng))和設備�;特殊情況下�,如采取特別防護措施并經(jīng)過必要的驗證,上述藥品制劑則可通過階段性生產(chǎn)方式共用同一生產(chǎn)設施和設備���。
• 上述這幾項的空氣凈化系統(tǒng),其排風應當經(jīng)過凈化處理(并且排風口應不能位于其他空調取風口的上游)���。
• 藥品生產(chǎn)廠房不得用于生產(chǎn)對藥品質量有不利影響的非藥用產(chǎn)品���。
適當?shù)那逑闯绦驅Ψ乐刮廴竞徒徊嫖廴揪哂兄匾饔茫鍧嵆绦蚴切枰A先經(jīng)過驗證的����。至少連續(xù)三次應用清洗程序,并都獲得成功���,才能證明該方法的有效性����。
清潔驗證的目的是證明設備對產(chǎn)品、洗滌劑和微生物殘留物的清洗一致并達到可接受的水平�,以防止可能的污染和交叉污染。清潔驗證被認為在多產(chǎn)品設施中尤為重要����,并應在設備、消毒程序和服裝洗滌等方面都進行驗證�。
對于難以清潔的產(chǎn)品、難以清潔的設備或安全風險高的產(chǎn)品���,如果使用經(jīng)過驗證的清潔程序無法達到所需清潔驗證的接受水平���,則應考慮使用專用的設備。
1.4工藝控制參數(shù)
一些工藝設備在驗證的時候需要考慮最差情況���,如隧道烘箱在做FH值驗證時有3種裝載形式:(1)履帶前面尚沒有瓶���,即開始生產(chǎn)的前期;(2)履帶式前后都有瓶,即生產(chǎn)的中間時段�;(3)履帶后面已沒有瓶,即生產(chǎn)將結束時���。在驗證中可以發(fā)現(xiàn)這3種裝載形式的FH值是不一樣的����,一般來說第二種裝載形式的FH值最高���。第一和第三種裝載的FH值哪個更高不能確定�,這與隧道烘箱內原設計的風平衡策略有關�。隧道烘箱的關鍵控制參數(shù)應該按照最低的FH值來設定,以滿足FH值最低時也能夠達到除熱源的目的���。
2、生產(chǎn)環(huán)境
2.1高效過濾器
無菌空氣是直接與物料相接觸的���,特別是在除菌過濾工藝以后�,如果不能夠確保無菌空氣的質量�,將給產(chǎn)品帶來巨大的風險。高效過濾器必須確保其完整性符合要求�,從而保障環(huán)境空氣的無菌效果。相關法規(guī)中規(guī)定高效過濾器的局部泄漏率要小于0.01%,并且對無菌制劑的生產(chǎn)設施來說應每半年就檢測一次�。當然,這一檢漏行為嚴格意義上講只能稱為完整性檢測�,因為這僅是用戶在使用狀態(tài)而非標準狀態(tài)下進行的檢測。
由于采用的標準不同以及對標準理解不透徹�,在檢漏過程中可能會存在著一定的風險。首先檢漏所選擇的儀器不能是激光粒子計數(shù)儀�,雖然激光粒子計數(shù)儀的靈敏度較高,但它僅能對一個粒徑的粒子進行計量�。所以它在高效過濾器下游計量到的粒子是不全面的,這樣檢測到的泄漏率自然就偏小了���。只有在用單分散粒徑的氣溶膠(如聚苯乙烯乳膠PSL)發(fā)塵時�,其檢測結果才與真實數(shù)據(jù)接近����。需要用光度計來進行高效過濾器下游粒子的計量,它是能夠計量所有透過粒徑粒子的�。
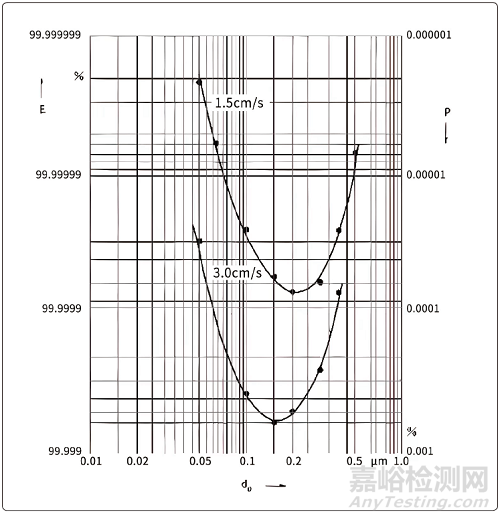
在保證高效過濾器上游的粒子濃度時,所用氣溶膠的粒徑也有很大的影響�,圖1是最易通過粒徑(MPPS)過濾效率的示意圖。舉例來說����,在采用PAO檢測時有冷發(fā)生和熱發(fā)生兩種發(fā)生器可供選擇。冷發(fā)生的粒徑大致為0.5?μm,熱發(fā)生的粒徑大致為0.3?μm���,從圖1上可以看到�,隨著粒徑增大����,過濾效率發(fā)生的變化很大。目前只有EN 1822標準才定義用MPPS來進行檢漏�,也只有用EN 1822標準進行檢測,檢測效率達到99.995%的高效過濾器才能稱為H14級別���。在EN 1822中有這樣一段話:對于H13級別過濾器(整體MPPS效率大于99.95%����,局部MPPS效率大于99.75%)���,在0.3?μm~0.5?μm粒徑的效率必須大于99.9996%(EN 1822-4:2009 Annex E.4 Leak criteria Page 40)�?��?梢娪捎诹降牟煌m然得到的檢測數(shù)據(jù)可能是相同的���,但實際上效率卻可能會相差一個甚至于幾個數(shù)量級���,這也是用戶檢漏數(shù)據(jù)會好于標準一個數(shù)量級的根本原因����。
圖1 MPPS示意圖
上文分析了高效過濾器過濾層的風險問題�,過濾層還有一個注意點:過濾風速(不同于過濾器的面風速)不應大于5?cm/s。
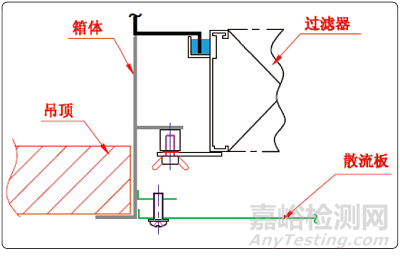
圖頂液槽的形式
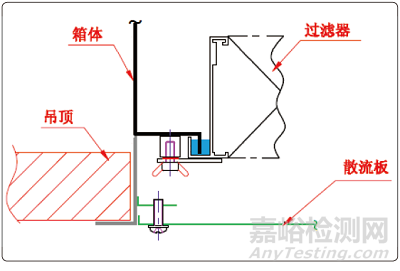
高效過濾器的安裝框架如果不采用液槽密封的形式�,邊框結合部也是很難滿足PAO泄露率小于0.01%的要求的。在高效過濾器與送風口的配合結構中�,有兩種形式:一種是頂液槽(如圖2所示)的形式,另一種是側液槽(如圖3所示)的形式����。這兩種結構的形式都能夠滿足PAO完整性檢測的需要,但頂液槽的形式并不適合用于無菌制劑的生產(chǎn)���。從圖2中可以清晰地看到�,高效過濾器與送風口之間有一個較大的死區(qū)域����,過濾器的風是循環(huán)不到該區(qū)域的。
圖3 側液槽的形式
2.2氣流組織
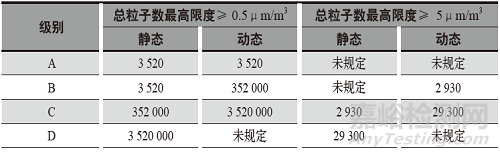
高效過濾器的完整性得到保障�,僅僅解決了高效過濾出風的質量問題���。不同的工藝操作還需要有相應的潔凈等級來支持,GMP對于不同的潔凈等級都有著嚴格的界定標準和判定指標����。各等級允許的總微粒限度,如表1所示�。
表1 各凈化級別允許的總微粒限度
A級:進行高風險操作的關鍵區(qū)域,通常情況下���,這種條件是通過局部氣流保護實現(xiàn)的����,應盡量減少操作人員對A 級區(qū)的直接干預���。
B級:對于無菌制備和灌裝來說�,這是A 級的背景潔凈室�。
C、D級:在無菌灌裝中執(zhí)行不太關鍵步驟操作的區(qū)域����,或作為隔離器的背景的潔凈室。
房間的自凈是通過連續(xù)換氣來達到的����。由高效過濾器吹出來的是無菌空氣,它不斷地置換和稀釋房間內的空氣�,從而確保房間滿足A、B����、C、D潔凈等級的需要�。不同潔凈等級的房間需要不同的換氣次數(shù),但房間的氣流組織流形比換氣次數(shù)更重要����。應盡量將房間設計成“頂送側下回”的流形,否則房間會存在換氣死角����,當然若能夠實現(xiàn)“頂送下回”是更為理想的。
不同級別的房間間會用壓差來避免交叉污染�,如若必要,相同級別的房間間也需要控制適當?shù)膲翰?���。但在這方面有一個誤區(qū),就是認為壓差越大越好���。其實并非如此�,一方面壓差升高,建筑圍護的風險就會增加���;另一方面�,也是更為重要的實際情況:壓差是滯留在房間內的氣體所生產(chǎn)的���,壓差越高滯留在房間內的氣體就越多���。這些滯留在房間內的氣體停留時間是不一樣的,不會像活塞流那樣齊刷刷地進出����,反而會影響房間的自凈效果。因此����,在做設計時,一旦發(fā)現(xiàn)核心區(qū)域的壓力很高����,就需要調整其壓力的分布情況,合理采用不同氣流形式的氣閘間����。
3����、質量回顧及趨勢分析
雖然無菌生產(chǎn)不太適合采用回顧性驗證�,但在一些關鍵數(shù)據(jù)上還是可以用統(tǒng)計分析來進行質量回顧和趨勢判斷�,從而評估污染和交叉污染的風險程度。本文接下來所講的統(tǒng)計學分析���,并不是簡單地畫USL(上規(guī)格限)�、LSL(下規(guī)格限)兩條上下限�,來查看運行數(shù)據(jù)是否落在之間。而是用工序能力指數(shù)來判斷可靠性如何���,將今年的可靠性與去年的可靠性進行比較���。用USL、LSL只能夠判斷所取樣品的數(shù)據(jù)情況���,用工序能力指數(shù)則可以預見未取樣數(shù)據(jù)的情況�,下面將列舉幾個例子進行說明����。
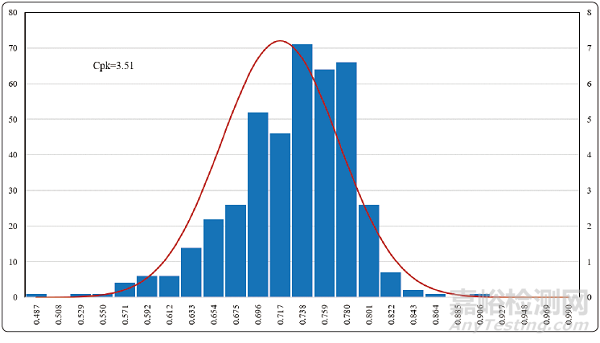
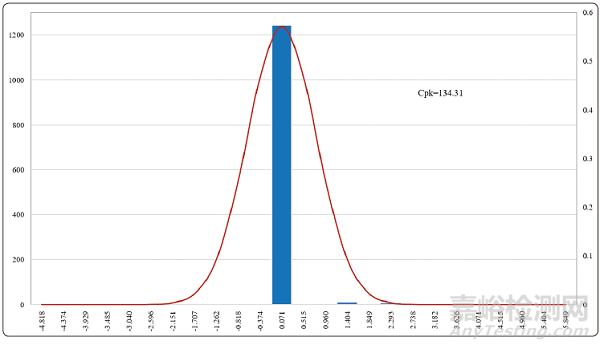
水系統(tǒng)方面已經(jīng)基本實現(xiàn)了電導率�、TOC(總有機碳)的在線監(jiān)測����,雖然微生物還是采用離線取樣的方法進行監(jiān)測,但對于這些監(jiān)測得到的數(shù)據(jù)完全可以采用工序能力指數(shù)的統(tǒng)計方法進行判斷和分析����。圖7-9是根據(jù)純化水系統(tǒng)一個完整年度的統(tǒng)計分析數(shù)據(jù)所做的控制圖。
圖4 純化水系統(tǒng)電導率控制圖
從圖4����、圖5和圖6的控制圖上可以清晰地看出,工序能力指數(shù)最小的電導率控制圖Cpk=3.51���,也就是說出現(xiàn)電導率超過1.3?μS/cm的概率為十億分之一����。
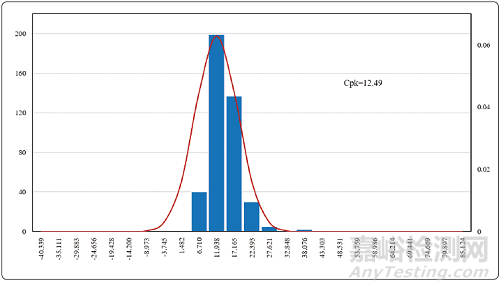
圖5 純化水系統(tǒng)TOC控制圖
圖6 純化水系統(tǒng)微生物控制圖
同樣道理���,滅菌柜在做Mapping的時候���,可以通過工序能力指數(shù)的方法來判斷潛在未布點的不達標可能性有多大�,干熱和濕熱滅菌柜都可以試用�。
對于無菌凍干粉針劑的生產(chǎn),也可以通過對成品中帶黑點不合格品的數(shù)量進行統(tǒng)計�,來對隧道烘箱高溫高效過濾器進行研判,以便提前發(fā)現(xiàn)高效過濾器泄漏的趨勢���。
4、結束語
本文從人���、機���、料、法���、環(huán)方面簡單地論述了污染可能存在的風險����,但不同的設施結構和產(chǎn)品有著獨特的情況���,需要進行針對性的分析和評價����。污染風險的識別和評估需要綜合考慮,有時在一個方面將其做到極致的時候�,在其他方面可能反而會出現(xiàn)問題。如上文提到的如果房間壓差控制過大�,會造成更多的氣流被滯留在室內,遠離了平推流的流形���,反而會削弱自凈的效率����。
對于一些有量化數(shù)據(jù)指標的場景����,可以采用工序能力指數(shù)來量化評判其風險的程度,這樣做比一些定性評估更有說服力���。一般認為靜態(tài)的驗證需要做三次����,這是為了考察其重現(xiàn)性���,如果用工序能力指數(shù)來評判就可以不必做滿三次����。
國家對于風險評估也有專門的技術規(guī)范:GB/T 27921-2011《風險管理 風險評估技術》,可以根據(jù)GB/T 27921-2011中推薦的方法來進行識別���、評估和應對�。在非最終滅菌無菌制劑的生產(chǎn)過程中���,經(jīng)常采用的風險評估技術為失效模式和后果分析����。它能夠很好地幫助企業(yè)化解識別的風險����,直至最終達到可接受的水平為止�。