隨著國(guó)內(nèi)IVD技術(shù)進(jìn)步及國(guó)家相關(guān)政策政策變化的推動(dòng),越來越多IVD公司積極開拓海外業(yè)務(wù)�����,布局全球市場(chǎng)�����。全球醫(yī)療器械的主要市場(chǎng)集中在美國(guó)��、歐洲��、日本����、中東、阿拉伯地區(qū)和中國(guó)�����,其中美國(guó)的醫(yī)療器械生產(chǎn)和消費(fèi)都占到幾乎全球的50%左右。中國(guó)醫(yī)療器械產(chǎn)品主要出口市場(chǎng)包含美國(guó)�����、歐盟�、英國(guó)、澳大利亞�、加拿大、韓國(guó)����、日本等。本文對(duì)各主要國(guó)家及地區(qū)醫(yī)療器械注冊(cè)要求及流程進(jìn)行了匯總�����。
美國(guó) FDA
市場(chǎng)價(jià)值:2021年�,全球醫(yī)械市場(chǎng)規(guī)模達(dá)5000多億美元。據(jù)媒體報(bào)道���,美國(guó)醫(yī)械市場(chǎng)占據(jù)了全球41%的市場(chǎng)份額�����。
FDA將醫(yī)療器械分為三類(Ⅰ����、Ⅱ�����、Ⅲ)�����,Ⅲ類風(fēng)險(xiǎn)管控等級(jí)高于Ⅰ�、Ⅱ類,這一點(diǎn)和國(guó)內(nèi)保持一致����。FDA將每一種醫(yī)療器械都明確規(guī)定其產(chǎn)品分類和管理要求,目前FDA對(duì)大約1700多種醫(yī)療器械產(chǎn)品進(jìn)行了分類涉及16個(gè)不同的版塊����。任何一種醫(yī)療器械想要進(jìn)入美國(guó)市場(chǎng),必須首先弄清申請(qǐng)上市產(chǎn)品分類和管理要求����。
步驟1:器械分類
如果想獲得 FDA 的正式器械確定或分類����,可向FDA提交 513(g) 請(qǐng)求��。但一般需要支付相應(yīng)的費(fèi)用�����。醫(yī)療器械依據(jù)其風(fēng)險(xiǎn)的程度��,分為以下3類:
Ⅰ類-低等風(fēng)險(xiǎn)(監(jiān)管控制類型:基本控制)大部分并不是所有�����,一般不需要510(K);
Ⅱ類-中等風(fēng)險(xiǎn)(監(jiān)管控制類型:基本控制以及特殊控制)大部分并不是所有�����,一般需要510(K);
Ⅲ類-高等風(fēng)險(xiǎn)(監(jiān)管控制類型:基本控制以及上市前批準(zhǔn))����。
步驟2:選擇正確的路徑遞交
器械分類確定之后,需要選擇相應(yīng)法規(guī)要求下的上市前遞交�。
常見的上市前遞交類型包括:
• 510(k)(上市前通知)
• PMA(上市前批準(zhǔn))
• De Novo(自動(dòng)III類指定的評(píng)價(jià))
• HDE (人道主義器械豁免)
一些Ⅰ類以及大部分Ⅱ類器械要求以510(k)的方式遞交。在510(k)遞交過程中����,申請(qǐng)者必須證明新的器械與對(duì)比器械在預(yù)期用途���,技術(shù)特征以及性能測(cè)試方面實(shí)質(zhì)等同。一些Ⅰ類和Ⅱ類器械可以豁免510(k)��,如果他們?cè)?1 CFR 862-892.9所述的豁免范圍之內(nèi)����。這些豁免被列在21 CFR的分類規(guī)則中�����,也被匯集在醫(yī)療器械豁免文件中����。
大部分Ⅲ類器械要求的遞交方式為PMA。PMA為嚴(yán)格程度較高的上市前遞交類型����。在FDA批準(zhǔn)PMA之前,申請(qǐng)者必須提供有效的科學(xué)證據(jù)����,以證明器械預(yù)期用途的安全性以及有效性����。
De Novo為沒有有效對(duì)比的新器械提供一種方式����,如果這種新器械滿足特定標(biāo)準(zhǔn),可以被分為Ⅰ或Ⅱ�����。
HDE為Ⅲ類器械提供了一種監(jiān)管路徑���,這類器械預(yù)期對(duì)罕見疾病或狀況的患者是有益的�。器械有資格成為人道主義豁免器械�,申請(qǐng)者必須獲得人道主義使用器械(HUD)的指定,可通過向FDA的孤兒產(chǎn)品開發(fā)辦公室Office of Orphan Products Development (OOPD)遞交申請(qǐng)���。
步驟3:準(zhǔn)備材料
在選擇正確的上市前遞交類型之后����,必須準(zhǔn)備該遞交類型所需的適當(dāng)?shù)馁Y料����。本部分將介紹在準(zhǔn)備上市前遞交時(shí)需要用到的有用資源以及考慮的信息���。FDA開發(fā)一些幫助申請(qǐng)者準(zhǔn)備上市前遞交的資源類型,包括:
器械建議(Device Advice)—綜合基于FDA上的網(wǎng)頁(yè)法規(guī)協(xié)助
510(k)的準(zhǔn)備��,參考:Premarket Notification 510(K)
PMA的準(zhǔn)備��,參考:Premarket Approval (PMA)
CDRH學(xué)習(xí)(CDRH Learn) —基于視頻的教學(xué)模塊���,研討會(huì)和錄制的包括各種政策和指導(dǎo)力度的網(wǎng)絡(luò)研討會(huì)
CDRH遞交前程序 —未來上市前提交申請(qǐng)可能要求FDA通過這個(gè)程序進(jìn)行反饋
準(zhǔn)備上市前遞交時(shí)需要考慮的信息:
設(shè)計(jì)控制:所有II類以及III類器械根據(jù)質(zhì)量管理體系(21 CFR 820.30)中對(duì)設(shè)計(jì)控制的要求進(jìn)行設(shè)計(jì)����。一些I類器械可豁免設(shè)計(jì)控制����。
非臨床測(cè)試:器械上市要求的測(cè)試以及信息類型是通過器械的分類,作用機(jī)制����,技術(shù)特征�����,以及標(biāo)簽來確定的�。醫(yī)療器械上市前遞交實(shí)施的非臨床測(cè)試必須符合21 CFR 58中的良好試驗(yàn)管理規(guī)范(GLPs)
臨床證據(jù):PMAs,HDEs 以及部分 510(k)s 和 De Novos 要求有臨床證據(jù)。在早起的臨床研究開始之前��,研究申請(qǐng)者需要得到FDA器械臨床研究豁免(IDE)的批準(zhǔn)����。這項(xiàng)研究也需要得到倫理審查委員會(huì)(IRB)的批準(zhǔn)。臨床研究必須符合所有的適用的器械臨床研究豁免(IDE)法規(guī)以及良好試驗(yàn)管理規(guī)范(GLPs)�����。
標(biāo)簽:器械的標(biāo)簽必須依據(jù)標(biāo)簽法規(guī)書寫,且需要包含在上市前遞交的資料中�����。
步驟4:遞交資料
提交給FDA���,并在FDA的工作人員審查過程中保持聯(lián)系�。
用戶費(fèi)用:在510(k)或PMA遞交時(shí)��,需要一定的用戶費(fèi)用
電子副本(eCopy):上市前遞交必須包含以光盤(CD)����、數(shù)字視頻光盤(DVD),或閃存驅(qū)動(dòng)器方式形成的電子副本�����。
行政備案審查:在上市前遞交接收之后����,F(xiàn)DA進(jìn)行行政審查��,評(píng)估遞交是否是足夠完整的�,以接收實(shí)質(zhì)性審查。
審查互動(dòng)(Interactive Review):當(dāng)遞交的資料處于正在審查中時(shí),F(xiàn)DA將和申請(qǐng)者保持聯(lián)系以增加審查過程中的效率�。
步驟5:完成登記
器械設(shè)備必須在FDA對(duì)其生產(chǎn)的企業(yè)進(jìn)行登記,并對(duì)其器械進(jìn)行列名�。如果一個(gè)器械在上市前需要上市前清關(guān)(premarket clearance)或上市前批準(zhǔn)(premarket approval),器械廠商在登記和列名之前必須等到它獲得FDA的清關(guān)或批準(zhǔn)���。器械企業(yè)登記�、登記號(hào)的分配或醫(yī)療器械的列名��,都不意味著FDA對(duì)其企業(yè)或其產(chǎn)品的清關(guān)或批準(zhǔn)����。
歐洲CE
市場(chǎng)價(jià)值:根據(jù)MedTec Europe統(tǒng)計(jì),2022歐洲醫(yī)療器械市場(chǎng)規(guī)模約為1350億歐元���,約占全球市場(chǎng)的27%�����,是僅次于美國(guó)的第二大醫(yī)療器械市場(chǎng)���。
自2021年5月起,醫(yī)療器械制造商必須遵守歐盟醫(yī)療器械法規(guī)2017/745而不是醫(yī)療器械指令93/42/EEC�����,才能獲得CE標(biāo)志批準(zhǔn)。因此����,醫(yī)療器械必須根據(jù)MDR進(jìn)行分類。但是�,一些分類規(guī)則保持不變,因此請(qǐng)隨意檢查以下MDD分類路線�。
如果您是制造商并且希望將您的醫(yī)療設(shè)備投放到歐盟市場(chǎng),您需要確保其符合歐盟委員會(huì)制定的特定歐洲指令���。在這種情況下�,重要的是醫(yī)療器械指令(MDD):AIMDD90/385/EEC�;MDD93/42/EEC�;IVDMDD98/79/EC。為了證明您的設(shè)備符合這些CE指令的基本要求�����,您需要在其上貼上CE標(biāo)志�。您的產(chǎn)品需要通過CE認(rèn)證標(biāo)記過程�����。后者的方向取決于您的醫(yī)療器械類別和您選擇的合格評(píng)定途徑。您的醫(yī)療設(shè)備的具體特性將決定其類別����,以及對(duì)患者的風(fēng)險(xiǎn)程度。例如,預(yù)期用途�����、侵入性以及局部與全身效應(yīng)等特征��。
根據(jù)歐洲框架�����,醫(yī)療器械分為四類:I類����、IIa類�����、IIb類和III類����。III類醫(yī)療器械的風(fēng)險(xiǎn)最高���。今天,由于新監(jiān)管系統(tǒng)的更嚴(yán)格規(guī)則��,許多設(shè)備的類別發(fā)生了變化。之前他們會(huì)被歸入IIa或IIb類��,但現(xiàn)在他們將被歸入III類����。如果您的醫(yī)療設(shè)備屬于I類以外的任何其他類別,您必須向認(rèn)證機(jī)構(gòu)提供證明����,證明您的產(chǎn)品符合相應(yīng)CE指令的基本要求。
第一類醫(yī)療器械CE
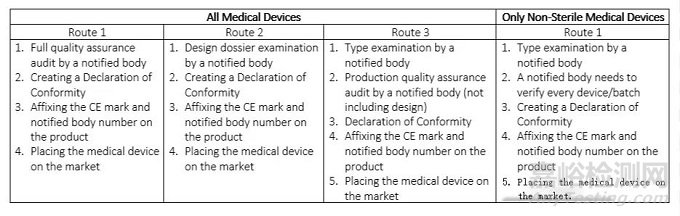
醫(yī)療器械類I的風(fēng)險(xiǎn)最低。此類設(shè)備的制造商可以選擇三種可能的CE標(biāo)記途徑中的一種�����。在這方面�,他們應(yīng)該考慮以下幾點(diǎn):如果醫(yī)療器械是否是無菌的,例如個(gè)人防護(hù)套件�;醫(yī)療器械是否具有測(cè)量功能,如聽診器�;如果它不是無菌的,也不是測(cè)量的���,例如矯正眼鏡�。注意:如果您的產(chǎn)品是I類產(chǎn)品����,并且它不是無菌或測(cè)量設(shè)備,那么您只需對(duì)其進(jìn)行自我認(rèn)證���,并通過書面聲明正式聲明其符合MDD的適用要求�����。如果它是無菌或測(cè)量醫(yī)療設(shè)備����,那么您將需要認(rèn)證機(jī)構(gòu)評(píng)估。

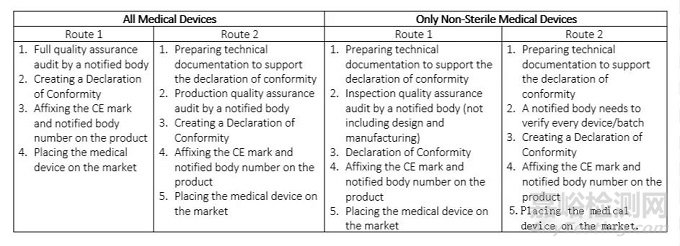
表1:I類醫(yī)療器械的CE認(rèn)證標(biāo)志路線
IIa類醫(yī)療器械CE
IIa類醫(yī)療器械可能是手術(shù)手套����、助聽器、診斷超聲機(jī)等�����。它們通常構(gòu)成中低風(fēng)險(xiǎn)�����?�;颊邞?yīng)短期使用���,不超過30天。如果您是IIa類醫(yī)療設(shè)備的制造商����,您必須支持您的聲明符合認(rèn)證機(jī)構(gòu)評(píng)估。只有這樣�,您才能將您的產(chǎn)品投放市場(chǎng)�����。有四種可能的途徑可以讓您的產(chǎn)品獲得CE標(biāo)志�����,根據(jù)產(chǎn)品的類型(即是否無菌)分為兩組�。
表2.IIa類醫(yī)療器械的CE標(biāo)志路線
IIb類醫(yī)療器械CE
在這里��,我們可以包括醫(yī)療設(shè)備��,例如長(zhǎng)期矯正隱形眼鏡��、手術(shù)激光���、除顫器等��。它們是中高風(fēng)險(xiǎn)設(shè)備�����,患者可能會(huì)使用它們超過30天�����。如果您的產(chǎn)品屬于IIb類��,類似于IIa類的程序�����,您將需要指定機(jī)構(gòu)來評(píng)估您的技術(shù)文檔是否符合醫(yī)療器械指令��。特定CE標(biāo)志路線的選擇將再次取決于您的產(chǎn)品類型�。
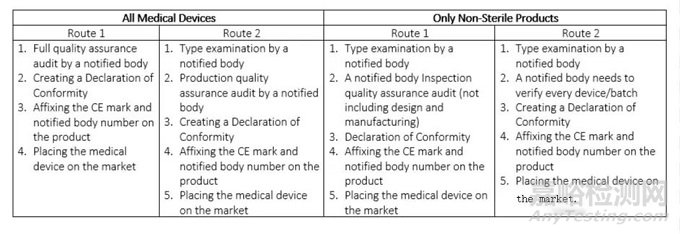
表3.IIb類醫(yī)療器械的CE標(biāo)志路線
第三類醫(yī)療器械CE
在該類別中,所有醫(yī)療設(shè)備都具有最高的風(fēng)險(xiǎn)�,并且需要在其生命周期內(nèi)進(jìn)行永久監(jiān)控。有專門的機(jī)構(gòu)負(fù)責(zé)對(duì)產(chǎn)品進(jìn)行監(jiān)測(cè)��。例如����,此類裝置是心血管導(dǎo)管、動(dòng)脈瘤夾�����、髖關(guān)節(jié)植入物、人工心臟瓣膜等�����。在這里,以及在II類中�����,醫(yī)療器械的合格評(píng)定可能包括對(duì)技術(shù)文件的審核和質(zhì)量體系/產(chǎn)品檢查�����,并側(cè)重于器械設(shè)計(jì)和生產(chǎn)的一個(gè)或多個(gè)方面。
表4三類醫(yī)療器械的CE標(biāo)志路線
英國(guó)UKCA
英國(guó)脫歐后�,UKCA 標(biāo)志于 2021 年 1 月在英國(guó)大不列顛 (Great Britain) 正式生效。一些分類的醫(yī)療器械可能需要持有 UKCA 認(rèn)證����。CE向UKCA的過渡期將持續(xù)至 2023年 6 月 30 日。
自 2021 年 1 月 1 日起���,無論是持有 UKCA 認(rèn)證或 CE 認(rèn)證的醫(yī)療器械�,在投放英國(guó)市場(chǎng)之前必須在 MHRA(Medicines and Healthcare products Regulatory Agency) 注冊(cè)��。
對(duì)于北愛爾蘭��,歐盟 MDR 和 IVDR 將分別于 2021 年 5 月 26 日和 2022 年 5 月 26 日起執(zhí)行�����。即使在 2023 年 7 月 1 日之后�����,在北愛爾蘭上市的醫(yī)療器械仍需持有 CE 標(biāo)志�����,制造商需要滿足歐盟法規(guī)��。
1. UKCA 標(biāo)志的標(biāo)簽和使用說明 (IFU) 要求
如果相關(guān)�,務(wù)必確保標(biāo)簽和 IFU 已更新,體現(xiàn) UKCA 標(biāo)志����。這方面的主要考慮因素包括:
標(biāo)簽必須體現(xiàn) UKCA 標(biāo)志,如果有授權(quán)機(jī)構(gòu)參與合格評(píng)定過程����,需包含該授權(quán)機(jī)構(gòu)編號(hào)。
對(duì)于非英國(guó)制造商����,如果使用 UKCA 標(biāo)志進(jìn)入英國(guó)大不列顛市場(chǎng) (Great Britain),需要在標(biāo)簽或外包裝或 IFU 上(取決于設(shè)備類型)上顯示英國(guó)授權(quán)代表 (UKRP) 的詳細(xì)信息����。IFU 需要體現(xiàn) UKCA 標(biāo)志。
2023 年 7 月 1 日之前�����,設(shè)備標(biāo)簽上可同時(shí)具有 CE 和 UKCA 標(biāo)志。2023 年 7 月 1 日之后��,英國(guó)大不列顛市場(chǎng) (Great Britain) 將繼續(xù)接受雙重標(biāo)志��。
2. 英國(guó)授權(quán)代表
如果制造商位于英國(guó)境外�����,必須指定一名英國(guó)授權(quán)代表 (UKRP)��。對(duì)于非英國(guó)制造商所有帶有 UKCA 或 CE 標(biāo)志的產(chǎn)品����,均必須符合這一要求。
UKCA合規(guī)過程中�����,F(xiàn)DASUNGO提供:
√ 英國(guó)合規(guī)負(fù)責(zé)人
√ 英國(guó)MHRA注冊(cè)申報(bào)
√ UKCA的技術(shù)文件更新或者編撰
√ 英國(guó)認(rèn)證機(jī)構(gòu)UKCA認(rèn)證評(píng)審輔導(dǎo)
√ 策劃應(yīng)對(duì)歐盟和英國(guó)市場(chǎng)準(zhǔn)入最優(yōu)方案
3. UKCA 標(biāo)志所需的符合性聲明 (DoC)
如果附上了 UKCA 標(biāo)志(包括器械帶有雙重標(biāo)志的情況)����,則您的 DoC 需要進(jìn)行 UKCA 標(biāo)志更新。DoC 應(yīng)體現(xiàn)英國(guó)相關(guān)法律要求����,包括引用的相關(guān)法律為 2002 年醫(yī)療器械法規(guī) (SI 618)��,及其 2019 (SI 791) 和 2020 年 (SI 1478) 的脫歐條例修正案�����。
4. 英國(guó)橫向立法和指定標(biāo)準(zhǔn)
作為 UKCA 的一部分,對(duì)橫向法律參考(Horizontal legislation references)進(jìn)行了變更�����。請(qǐng)參考圖表評(píng)估您的要求����。
此外,英國(guó)脫歐后�����,歐盟官方公報(bào)上公布的協(xié)調(diào)標(biāo)準(zhǔn)對(duì)其不再適用����。因此,英國(guó)也已公布了支持本國(guó)法規(guī)的標(biāo)準(zhǔn)清單�����。這些標(biāo)準(zhǔn)被稱為“指定標(biāo)準(zhǔn)designated standards”。
制造商若使用這些標(biāo)準(zhǔn)來證明其器械合規(guī)性�����,應(yīng)了解英國(guó)指定標(biāo)準(zhǔn)清單的公布情況��,并時(shí)刻關(guān)注有關(guān)這些清單更新的更多信息��。
5. 所有醫(yī)療器械都需要在MHRA登記注冊(cè)嗎����?包括體外診斷設(shè)備嗎?
是的��,所有醫(yī)療器械包括體外診斷設(shè)備都需要在英國(guó)MHRA進(jìn)行登記注冊(cè)并且有指定的英代(UKRP)才能在英國(guó)市場(chǎng)進(jìn)行銷售�����。
6. UKCA 標(biāo)志是否和CE 標(biāo)志一樣����,需要包含UK AB號(hào)?
根據(jù)UK法規(guī)的要求��,如果產(chǎn)品需要英國(guó)批準(zhǔn)機(jī)構(gòu)做符合性評(píng)估的話��,UKCA標(biāo)志是需要包含批準(zhǔn)機(jī)構(gòu)號(hào)碼的。
7. UKCA對(duì)于技術(shù)文檔有特殊的要求嗎�����?歐盟MDR的技術(shù)文檔是否可以滿足UKCA的要求�����?
一般來講��,MDR的要求是可以覆蓋UKCA的要求�����,因?yàn)閁KCA的要求是基于MDD�����。但是����,企業(yè)能夠熟悉不同法規(guī)對(duì)應(yīng)的不同要求是很重要的�����。
8. I類產(chǎn)品進(jìn)入英國(guó)市場(chǎng)可以自我宣稱嗎?需要獲得UKCA證書嗎����?
依據(jù)UK MDR 2002法規(guī)要求,I類產(chǎn)品和others類IVD產(chǎn)品在貼上UKCA 標(biāo)志和進(jìn)入英國(guó)市場(chǎng)前需要有自我宣稱的符合性聲明�����。但I(xiàn)類滅菌產(chǎn)品和I類帶測(cè)量功能產(chǎn)品在貼UKCA標(biāo)志和進(jìn)入英國(guó)市場(chǎng)前��,需要獲得UK AB的批準(zhǔn)��。
9. 愛爾蘭共和國(guó)是遵守UK MDR 2002��,還是EU MDR��?
愛爾蘭共和國(guó)是歐盟的一部分,進(jìn)入愛爾蘭市場(chǎng)需要遵守EU MDR。
澳洲TGA
TGA 對(duì)醫(yī)療器械的分類與歐盟幾乎一致�����,且歐盟 CE 是可以被 TGA 認(rèn)可的����,并可以作為滿足澳大利亞安全法規(guī)的重要注冊(cè)資料。
為了高效監(jiān)管��,醫(yī)療器械被TGA按照風(fēng)險(xiǎn)級(jí)別分為5類�����。分別是:
Class I. (低風(fēng)險(xiǎn))
Class IIa. (中低風(fēng)險(xiǎn))
Class IIb. (中高風(fēng)險(xiǎn))
Class III. (高風(fēng)險(xiǎn))
有源植入式醫(yī)療器械(AIMD).
這5類不包含體外診斷設(shè)備(IVD)��,體外診斷設(shè)備有獨(dú)立的分類程序����。
在確認(rèn)產(chǎn)品的醫(yī)療器械分類后��,申請(qǐng)人(在TGA認(rèn)證中被稱為Sponsors)應(yīng)當(dāng)提交產(chǎn)品的技術(shù)文檔給TGA����,TGA將花費(fèi)約200天完成審查(Class II),審查通過則簽發(fā)TGA證書����。
技術(shù)文檔應(yīng)當(dāng)包括產(chǎn)品測(cè)試報(bào)告��、技術(shù)文件以及制造商的質(zhì)量體系(QMS)等��。
澳大利亞TGA有條件認(rèn)可歐盟CE MDR�����,如果申請(qǐng)人提供有效的CE證書����,將能夠用于替代部分技術(shù)文件����。
同時(shí),TGA認(rèn)可MDSAP����,如果制造商能夠提供有效的MDSAP的審核報(bào)告,那么可以免于TGA對(duì)制造商質(zhì)量體系的審核��。
在TGA認(rèn)證完成后����,申請(qǐng)人將獲得TGA頒發(fā)的證書。
TGA證書長(zhǎng)期有效��,但是每年需要繳納年費(fèi)。
申請(qǐng)人必須是澳大利亞當(dāng)?shù)毓?���;如果?guó)外公司出口醫(yī)療器械到澳大利亞,則必須有當(dāng)?shù)毓咀鳛榉ㄒ?guī)聯(lián)系人(也被稱為當(dāng)?shù)卮恚?/span>
澳大利亞醫(yī)療用品管理局(TGA)也是TGA認(rèn)證的監(jiān)管機(jī)構(gòu)��,對(duì)于違反《醫(yī)療用品法》的行為��,最高可處罰5年監(jiān)禁和5000個(gè)罰款單位(約合350萬RMB)��。
品牌商應(yīng)當(dāng)謹(jǐn)慎保證產(chǎn)品合規(guī)�����。
加拿大CMDCAS
不同于美國(guó)����,亦不同于歐洲��,加拿大實(shí)行政府注冊(cè)結(jié)合第三方的質(zhì)量體系審查��。這里所說的第三方����,指經(jīng)加拿大標(biāo)準(zhǔn)委員會(huì) (SCC) 所認(rèn)可的能夠進(jìn)行加拿大醫(yī)療器械合格評(píng)定體系審核的第三方機(jī)構(gòu)��。
1����、不同于美國(guó)食品藥品管理局(FDA)政府一手抓到底即政府的產(chǎn)品注冊(cè)加上政府的現(xiàn)場(chǎng)審查(GMP審查)�����,亦不同于歐洲的完全第三方公告機(jī)構(gòu)(NotifiedBody)檢查制度(CE認(rèn)證)����,加拿大實(shí)行政府注冊(cè)結(jié)合第三方的質(zhì)量體系審查。這里所說的第三方����,指經(jīng)加拿大醫(yī)療器械認(rèn)證認(rèn)可機(jī)構(gòu)(CMDCAS,CanadianMedicalDevicesConformity)認(rèn)可的第三方機(jī)構(gòu),以下稱CMDCAS認(rèn)可機(jī)構(gòu)�����。
2����、加拿大醫(yī)療器械法規(guī)依據(jù)器械的使用風(fēng)險(xiǎn)將醫(yī)療器械分為I,II,III����,IV四個(gè)分類,如I類器械為最低風(fēng)險(xiǎn)��,IV類器械風(fēng)險(xiǎn)為最高����。為此針對(duì)制造者提出的產(chǎn)品注冊(cè)要求也是逐級(jí)增加,要求制造者實(shí)行的體系是愈加詳盡��。
審核點(diǎn)
對(duì)I類醫(yī)療器械(包括IVD)沒有質(zhì)量體系要求�����,并豁免產(chǎn)品注冊(cè)許可����。
加拿大醫(yī)療器械法規(guī)(Canadian Medical Device Regulations, CMDR)不要求進(jìn)口商或分銷商進(jìn)行質(zhì)量體系注冊(cè)。
加拿大的現(xiàn)行質(zhì)量體系標(biāo)準(zhǔn)為CAN/CSA-ISO 13485-03(R2013)����。
II類醫(yī)療器械制造商應(yīng)符合中除設(shè)計(jì)以外的要求;III和IV類器械應(yīng)符合包括設(shè)計(jì)在內(nèi)的所有CAN/CSA-ISO 13485-03(R2013)要求��。
質(zhì)量體系認(rèn)證由被第三方機(jī)構(gòu)簽發(fā),這些第三方機(jī)構(gòu)是由Standard Council of Canada(SCC)和加拿大衛(wèi)生部聯(lián)合指派的醫(yī)療器械符合性評(píng)估系統(tǒng)內(nèi)(Canadian Medical Devices Conformity Assessment System, CMDCAS)的機(jī)構(gòu)��。目前我國(guó)國(guó)內(nèi)TUV萊茵�����、TUV SUD�����、SGS等機(jī)構(gòu)具有CMDCAS資質(zhì)��。
與歐美醫(yī)療器械認(rèn)證方式不同的是��,加拿大CMDCAS認(rèn)證����,僅質(zhì)量體系部分由加拿大認(rèn)可的第三方機(jī)構(gòu)進(jìn)行審核,注冊(cè)文件的最終審核由加拿大衛(wèi)生部執(zhí)行�����。
韓國(guó) KFDA
韓國(guó)醫(yī)療器械法把醫(yī)療器械分為 4 類(Ⅰ�����、Ⅱ、Ⅲ����、Ⅳ),這種分類方法與歐盟對(duì)醫(yī)療器械的分類方法非常相似����。
韓國(guó)MFDS認(rèn)證把醫(yī)療器械按照風(fēng)險(xiǎn)級(jí)別分為四類:(不含體外診斷設(shè)備)
Class I. 極低風(fēng)險(xiǎn)產(chǎn)品。如眼科顯微鏡��、防輻射手套�����、手術(shù)臺(tái)����、聽診器等。目前有521個(gè)類別�����。
Class II. 低風(fēng)險(xiǎn)產(chǎn)品��。如脈搏血氧儀����、磁共振成像儀、腦電波檢測(cè)儀等�����。目前有1017個(gè)類別�����。
Class III. 中等風(fēng)險(xiǎn)產(chǎn)品�����。如麻醉系統(tǒng)�����、避孕套�����、縫合線等�����。目前有318個(gè)類別。
Class IV. 高風(fēng)險(xiǎn)產(chǎn)品����。如植入式除顫器、冠狀動(dòng)脈支架��、可生物降解椎間盤����、人工晶體等。目前有253個(gè)類別����。
體外診斷設(shè)備(IVD)有獨(dú)立的分類,目前也是分為4類��,一共有225種�����。
MFDS認(rèn)證必須由韓國(guó)當(dāng)?shù)毓景l(fā)起申請(qǐng)��,其申請(qǐng)流程和中國(guó)NMPA類似����,申請(qǐng)人需要提交產(chǎn)品的測(cè)試報(bào)告��、技術(shù)文件����、臨床報(bào)告及質(zhì)量體系證明等給相應(yīng)機(jī)構(gòu)�����,在獲得批準(zhǔn)后簽發(fā)MFDS證書����。
其中質(zhì)量體系(QMS)需要符合韓國(guó)良好生產(chǎn)規(guī)范(KGMP)����,制造商必須在經(jīng)過韓國(guó)MFDS認(rèn)可的機(jī)構(gòu)現(xiàn)場(chǎng)審核后才能獲得KGMP證書。
而測(cè)試必須在韓國(guó)MFDS認(rèn)可的實(shí)驗(yàn)室進(jìn)行�����,雖然目前MFDS有國(guó)外實(shí)驗(yàn)室認(rèn)可計(jì)劃����,但是醫(yī)療器械測(cè)試實(shí)驗(yàn)室目前都位于韓國(guó)本土。
具體來說�����,Class I類設(shè)備適用于通知程序,Class II類設(shè)備適用于認(rèn)證和批準(zhǔn)程序�����,Class III和Class IV類設(shè)備僅適用于批準(zhǔn)程序��。
一般情況下��,現(xiàn)存的Class I和Class II類設(shè)備由醫(yī)療器械信息和技術(shù)援助中心 (MDITAC)及韓國(guó)國(guó)家醫(yī)療器械安全信息研究所 (NIDS)認(rèn)證��;新產(chǎn)品以及Class III����、Class IV類設(shè)備則由MFDS直接審核。
醫(yī)療器械和人體健康安全息息相關(guān)�����,品牌商應(yīng)當(dāng)嚴(yán)格遵守《醫(yī)療器械法》《醫(yī)療器械法執(zhí)行條例》等法規(guī)��。
一旦被發(fā)現(xiàn)有違法行為����,會(huì)被按例處罰最高3000萬韓元(約合30萬RMB)�����。
日本 PMDA
醫(yī)療器械公司希望把產(chǎn)品投放到日本市場(chǎng)必須要滿足日本的 PMDA�����,但是語言問題和復(fù)雜的認(rèn)證程序還是日本醫(yī)療器械注冊(cè)的一個(gè)困難點(diǎn)����。
PMDA和MHLW
日本的藥品和醫(yī)療器械管理是由衛(wèi)生勞動(dòng)和福利部(日本厚生省)(Ministryof Health, Labor and Welfare����,MHLW)負(fù)責(zé)����。
PMDA全稱為Pharmaceuticals and Medical Devices Agency(獨(dú)立行政法人藥品和醫(yī)療器械綜合機(jī)構(gòu)),是MHLW管轄的獨(dú)立行政法人�����。PMDA的業(yè)務(wù)包括審查�����、安全對(duì)策、健康損害救濟(jì)等��,對(duì)醫(yī)療器械進(jìn)行技術(shù)復(fù)核和相關(guān)研究工作����。
在日本,藥品�����、醫(yī)療器械管理法律法規(guī)主要分為三類:
● 由日本議會(huì)批準(zhǔn)通過的稱法律��;
● 由日本政府內(nèi)閣批準(zhǔn)通過的稱政令或法令��;
● 由厚生省大臣批準(zhǔn)通過的稱告示或省令��。
注冊(cè)流程詳解
1. 確定產(chǎn)品分類
日本醫(yī)療器械術(shù)語集(Japanese Medical Device Nomenclature , JMDN) 編碼明確了器械分類與注冊(cè)登記路徑�����。根據(jù)醫(yī)療器械的風(fēng)險(xiǎn)等級(jí)�����,分為一類、二類�����、三類和四類��。
● 一類為一般醫(yī)療設(shè)備��,認(rèn)為即使發(fā)生不良事件����,對(duì)人體的風(fēng)險(xiǎn)也極低的產(chǎn)品,如手術(shù)刀等��。→ 須進(jìn)行地方政府備案�����,無實(shí)質(zhì)性審查����。
● 二類為管制醫(yī)療設(shè)備��,認(rèn)為即使發(fā)生不良事件��,對(duì)人體的風(fēng)險(xiǎn)也比較低的產(chǎn)品,如電子內(nèi)窺鏡��、消化器官用導(dǎo)管等����。→ 須由第三方認(rèn)證機(jī)構(gòu)RCB負(fù)責(zé)審查。
● 三類為高度管制醫(yī)療設(shè)備����,認(rèn)為在發(fā)生不良事件時(shí),對(duì)人體的風(fēng)險(xiǎn)比較高的產(chǎn)品��。如透析器��、人工骨骼�����、人工呼吸器��、心臟血管用球囊導(dǎo)管等����,→ 須進(jìn)行PMDA審查
● 四類為高度管制醫(yī)療設(shè)備,是對(duì)患者的侵入性高����、在發(fā)生不良事件的情況下有可能直接導(dǎo)致生命危險(xiǎn)的產(chǎn)品�����,如起搏器�����、人工心臟����、支架等�����。→ 須進(jìn)行PMDA審查
2. 任命MAH/D-MAH
所有類別器械:任命MAH或D-MAH管理日本器械上市前申請(qǐng)或?qū)徟?/span>
MAH全稱Marketing Authorized Holder(日本上市許可持有人)�����,拿到某一類產(chǎn)品MAH執(zhí)照后�����,才可以提出具體產(chǎn)品的上市申請(qǐng)�����。由于外國(guó)公司在日本沒有辦事處�����,需要任命一名在日本持有營(yíng)業(yè)執(zhí)照的指定上市許可持有人D-MAH (Designated Marketing Authorization)��,協(xié)調(diào)貨物放行給外國(guó)公司的經(jīng)銷商以及處理投訴和警戒信息事宜��。
3. 進(jìn)行制造商登記
● 日本制造商向地方當(dāng)局提交制造商注冊(cè) (MR) 申請(qǐng)�����。
● 外國(guó)制造商向PMDA提交外國(guó)制造商注冊(cè) (FMR) 申請(qǐng)����。
醫(yī)療器械產(chǎn)品投放到日本市場(chǎng)必須滿足日本藥品和醫(yī)療器械法案(Pharmaceutical and Medical Device Act, PMD Act)。
● PMD Act要求日本本國(guó)制造商向當(dāng)?shù)貦C(jī)構(gòu)申請(qǐng)其生產(chǎn)制造場(chǎng)所的注冊(cè)登記����,并獲得制造商注冊(cè)登記(Manufacturer registration, MR) 證書;
● PMD Act要求外國(guó)制造商向PMDA申請(qǐng)其生產(chǎn)制造場(chǎng)的注冊(cè)登記�����,并獲得外國(guó)制造商注冊(cè)登記(Foreign manufacturer registration, FMR)證書。
● MR和FMR證書是提交醫(yī)療器械注冊(cè)登記申請(qǐng)時(shí)的一項(xiàng)要求��,提出申請(qǐng)前必須取得證書�����。
4. 質(zhì)量管理體系 J-GMP
(MHLW Ordinances No.169)
● 一類器械不需要J-GMP審核��。
● 二類器械由注冊(cè)認(rèn)證機(jī)構(gòu)(RCB)進(jìn)行J-GMP審核�����。
● 二類(除特殊控制外)��、三類和四類器械����,由PMDA進(jìn)行QMS審核。
● 沒有JMDN編碼的新器械����、四類器械或需要進(jìn)行臨床試驗(yàn)的器械,通常會(huì)進(jìn)行現(xiàn)場(chǎng)審查��。
● J-GMP是進(jìn)入日本市場(chǎng)的醫(yī)療器械企業(yè)必須滿足的����、基于ISO 13485 并附加日本法律法規(guī)特殊要求的一套質(zhì)量管理體系要求。
5. 提交上市申請(qǐng)
1. 上市前注冊(cè)申請(qǐng)(Todokede)
一類醫(yī)療器械(必須向PMDA提交一份上市前注冊(cè)申請(qǐng)��。申請(qǐng)為通告性文件��,PMDA不會(huì)做出任何審查意見����。
2. 上市前認(rèn)證(Ninsho)
有認(rèn)證標(biāo)準(zhǔn)(JapaneseIndustrial Standards, JIS)的二類(及部分三類)器械必須通過上市前認(rèn)證。MAH向注冊(cè)認(rèn)證機(jī)構(gòu)(RCB)提交申請(qǐng)并通過認(rèn)證��。
3. 上市前審批(Shonin)
除了特殊控制的二類器械外的其他二類器械和三����、四類器械,由MAH或DMAH向PMDA提交上市前審批申請(qǐng)�����,并獲得厚生勞動(dòng)省(MHLW)的批準(zhǔn)��。
6. 頒發(fā)證書
● 二類器械由RCB頒發(fā)上市前認(rèn)證證書����。
● 二類(除特殊控制外)�����、三類和四類器械由MHLW頒發(fā)上市前批準(zhǔn)證書����。
● 器械注冊(cè)無有效期�����。
新加坡HAS
在新加坡��,醫(yī)療器械產(chǎn)品分為A��、B����、C、D等4類�����。
A類醫(yī)療器械為低風(fēng)險(xiǎn)產(chǎn)品��,如醫(yī)用擴(kuò)張器/壓舌板;B類為中低風(fēng)險(xiǎn)產(chǎn)品,如皮下注射針/吸入式設(shè)備;C類為中高風(fēng)險(xiǎn)產(chǎn)品��,如肺通氣器/接骨板;D類為高風(fēng)險(xiǎn)產(chǎn)品�����,如心臟瓣膜/可植入除顫器����。
A類醫(yī)療器械的注冊(cè)申請(qǐng)分為4步��,即提交申請(qǐng)�����、篩選�����、評(píng)審����、主管部門作出決定?����! ?/span>
B、C����、D類產(chǎn)品的申請(qǐng)材料包括,參照東盟通用提交資料模板(CSDT)準(zhǔn)備的英文申請(qǐng)��,相關(guān)證書����、報(bào)告和標(biāo)簽復(fù)印件。同時(shí)�����,還應(yīng)提交良好的流通規(guī)范證書(GDPMDS)或醫(yī)療器械質(zhì)量管理體系ISO 13485證書��。申請(qǐng)公司應(yīng)指定一名主要聯(lián)系人����,負(fù)責(zé)與主管部門聯(lián)絡(luò)與申請(qǐng)相關(guān)的所有問題,包括按要求補(bǔ)充材料�����。。
俄羅斯
主管當(dāng)局:
俄羅斯聯(lián)邦居民健康與社會(huì)發(fā)展監(jiān)督部(RZN)
要求及流程:
1.確定分類�����,并確定命名分類代碼��。
2.任命一名授權(quán)代表來協(xié)調(diào)您在俄羅斯的醫(yī)療器械注冊(cè)�����。
3. 將設(shè)器械信息和現(xiàn)有測(cè)試報(bào)告發(fā)送到俄羅斯的授權(quán)測(cè)試實(shí)驗(yàn)室�����,文件必須翻譯成俄文��。
4. 準(zhǔn)備并向 RZN 提交進(jìn)口許可申請(qǐng)����, RZN 頒發(fā)許可證��。
5. 將器械發(fā)送到俄羅斯的授權(quán)測(cè)試實(shí)驗(yàn)室進(jìn)行質(zhì)量����、安全和功效測(cè)試。
6. 準(zhǔn)備注冊(cè)文檔,包括技術(shù)信息��、測(cè)試結(jié)果����、ISO 13485 證書和現(xiàn)有臨床數(shù)據(jù)。提交給 RZN 并支付費(fèi)用��。大多數(shù)文件必須翻譯成俄文�����。證書必須經(jīng)過公證和加注
7. 對(duì)于 IIa����、IIb 和 III 類:RZN 進(jìn)行完整性審查。如果可以接受����,檔案將發(fā)送到專業(yè)技術(shù)中心進(jìn)行技術(shù)審查。專業(yè)知識(shí)中心審查補(bǔ)充臨床數(shù)據(jù)要求并提出建議��。RZN 審查建議并就后續(xù)步驟發(fā)表最終意見��。
8. 對(duì)于 IIa����、IIb 和 III 類:按照 RZN 的臨床數(shù)據(jù)要求��,在俄羅斯進(jìn)行額外的測(cè)試或試驗(yàn)����。
9. 對(duì)于所有器械類別:文檔經(jīng)過第 2 階段審查��。如果可以接受��,RZN 將頒發(fā)注冊(cè)證書并在 RZN 網(wǎng)站的注冊(cè)數(shù)據(jù)庫(kù)中列出產(chǎn)品�����。注冊(cè)不會(huì)過期��。
10.對(duì)于所有器械類別:指定俄羅斯聲明人并申請(qǐng)符合性聲明 (DoC) 認(rèn)證����。
巴西
主管當(dāng)局:
ANVISA全稱Agência Nacional de Vigilancia Sanitária��,隸屬巴西衛(wèi)生部����,負(fù)責(zé)所有醫(yī)療器械��、體外診斷產(chǎn)品及其他健康相關(guān)產(chǎn)品(如藥品��、衛(wèi)生用品�����、化妝品等)的上市前與上市后的管控�����。
要求及流程:
巴西醫(yī)療器械分類:ANVISA將醫(yī)療設(shè)備(medical equipments)�����,用于健康的材料(materials for health use)����,骨科植入物(orthopedic implants)和體外診斷(in vitro diagnostics)統(tǒng)稱為醫(yī)療器械��。
巴西的醫(yī)療器械應(yīng)根據(jù)其對(duì)消費(fèi)者�����、患者��、經(jīng)營(yíng)者或相關(guān)第三方構(gòu)成的風(fēng)險(xiǎn)程度,將其劃分為第I�����、II����、III或IV類。
對(duì)于 II�����,III或IV類醫(yī)療器械�����,應(yīng)提交的注冊(cè)資料如:
a)相應(yīng)的健康監(jiān)督費(fèi)的支付憑證��;
b)用于識(shí)別制造商或進(jìn)口商及其醫(yī)療器械的信息��;
c)海外制造商或出口商授權(quán)進(jìn)口商在巴西將該醫(yī)療器械商業(yè)化的授權(quán)書副本��。經(jīng)出口經(jīng)營(yíng)者授權(quán)��,進(jìn)口經(jīng)營(yíng)者應(yīng)當(dāng)說明生產(chǎn)經(jīng)營(yíng)者與出口經(jīng)營(yíng)者之間的商業(yè)關(guān)系�����;
d)進(jìn)口醫(yī)療器械��,由醫(yī)療器械生產(chǎn)和營(yíng)銷所在國(guó)主管當(dāng)局頒發(fā)的注冊(cè)證明或免費(fèi)銷售證明(或同等文件)��;
e)證明符合技術(shù)法規(guī)中所載的法律條文�����,例如管制醫(yī)療產(chǎn)品的ANVISA法例��。
I類醫(yī)療器械注冊(cè)的制造商或進(jìn)口商����,須向ANVISA提交上述第(a)、(b)及(e)項(xiàng)所列明的文件����。
有源類醫(yī)療器械還需要先進(jìn)行INMETRO認(rèn)證,再提交ANVISA進(jìn)行注冊(cè)����。
巴西醫(yī)療器械注冊(cè)對(duì)于體系的要求:
GMP證書是ANVISA發(fā)行的證明企業(yè)符合良好生產(chǎn)規(guī)范的證書。出口到巴西的III,IV類醫(yī)療器械����,需要符合BGMP的要求�����。
對(duì)于醫(yī)療器械�����,國(guó)際檢查僅適用于制造III級(jí)和IV級(jí)風(fēng)險(xiǎn)產(chǎn)品的公司�����。外國(guó)申請(qǐng)人公司的巴西代表必須每?jī)赡旮乱淮蜧MP證書�����。但是�����,將進(jìn)行風(fēng)險(xiǎn)分析,以決定是否需要重新檢查或是否可以根據(jù)文件分析來更新證書�����。
阿根廷
主管當(dāng)局:
國(guó)家藥品、食品和醫(yī)療技術(shù)管理局(ANMAT)
要求及流程:
根據(jù)ANMAT的要求�����,申請(qǐng)批準(zhǔn)的醫(yī)療器械需要滿足以下要求:
1.指定阿根廷授權(quán)代表(AAR):ANMAT要求尋求授權(quán)的醫(yī)療器械公司必須指定一個(gè)當(dāng)?shù)卮碜鳛檎麄€(gè)注冊(cè)過程的聯(lián)絡(luò)人��。AAR必須獲得ANMAT良好生產(chǎn)規(guī)范(GMP)認(rèn)證����,涵蓋提交的設(shè)備,并持有授權(quán)許可�����。對(duì)于II����、III、IV類器械以及IVD�����,AAR需要提交:
2.自由銷售證書(CFS)或給外國(guó)政府的證書(CFG)
3.商業(yè)歷史
4.檔案資料
5.注冊(cè)費(fèi)支付證明
6.南方共同市場(chǎng)符合性聲明
7.報(bào)告召回以及現(xiàn)場(chǎng)安全糾正措施的宣誓書
注:對(duì)于I類器械�����,AAR只需要提交付款證明、制造商信息和南方共同市場(chǎng)符合性聲明����。
8.自由銷售證書(CFS)/外國(guó)政府證書(CFG):CFS或CFG必須由與ANMAT有協(xié)議的國(guó)家的認(rèn)可機(jī)構(gòu)(擁有醫(yī)療設(shè)備及其適用配件的信息,以及制造商的名稱)提供�����。
9.西班牙語翻譯的文件:所有提交給ANMAT的文件必須翻譯成西班牙語�����。此外����,還必須有:設(shè)備分類、使用說明(IFU)�����、標(biāo)簽�����、制造商的信息和技術(shù)文件��。
墨西哥
主管當(dāng)局:
防御衛(wèi)生風(fēng)險(xiǎn)聯(lián)邦委員會(huì)(COFEPRIS)
要求及流程:
1.委任一名墨西哥注冊(cè)持有人(MRH)來作當(dāng)?shù)卮怼?/span>
2.對(duì)證明器械獲得了原產(chǎn)國(guó)的批準(zhǔn)�����。* 滿足這一要求的常用方法是使用自由銷售證書(CFS�����,Certificate of Free Sale)或 致外國(guó)政府函(CFG��,Certificate to Foreign Government)��。
3.指定一名具有資質(zhì)的經(jīng)銷商將您的醫(yī)療器械或IVD進(jìn)入墨西哥�����。
4.對(duì)于I類低風(fēng)險(xiǎn)醫(yī)療器械:向COFEPRIS提交包括公司和醫(yī)療器械基本信息在內(nèi)的申請(qǐng)�����。所有文件必須用西班牙語提交����。
對(duì)于I、II和III類器械:編制詳細(xì)的注冊(cè)檔案,包括器械/制造信息�����、實(shí)驗(yàn)室測(cè)試報(bào)告等��。提供符合質(zhì)量管理要求的證據(jù)(例如ISO 13485證書)和/或CE證書��。
5.對(duì)于I����、II和III類器械:提供特定的測(cè)試報(bào)告,具體取決于產(chǎn)品的功能和預(yù)期用途�����。
6.對(duì)于所有類別:所有器械必須遵循NOM-137-SSA1-2008的標(biāo)簽要求��。標(biāo)簽和說明書必須使用西班牙語�����。
7.對(duì)于I�����、II和III類器械:MRH向COFEPRIS或第三方審查人**提交注冊(cè)檔案以供審查,并支付注冊(cè)費(fèi)用����。所有文件必須用西班牙語提交����。
8.對(duì)于所有類別:COFEPRIS頒發(fā)證書并在COFEPRIS網(wǎng)站上發(fā)布注冊(cè)登記證明。注冊(cè)證書的有效期為5年����。有些產(chǎn)品可能需要進(jìn)口許可證才能進(jìn)入墨西哥。
其它國(guó)家醫(yī)療器械和IVD注冊(cè)介紹
巴拿馬����、秘魯、阿拉伯�����、泰國(guó)��、越南����、印尼�����、哈薩克�����、尼泊爾�����、巴基斯坦��、斯里蘭卡�����、意大利�����、波蘭��、德國(guó)�����、巴爾干����、維也納、埃及����、蘇丹�����、西非��、沙特�����、迪拜��、波斯��、伊朗����、利比亞��,可以直接做CE的注冊(cè)就能搞定基本所有國(guó)家����。但根據(jù)產(chǎn)品分類不同流程有所區(qū)別����。
I類的只要自我申明符合MDD指令要求就可以,III類非常繁瑣�����,需要所有技術(shù)文件交所在國(guó)(一般是德國(guó))主管當(dāng)局審核����。其他所有國(guó)家,屬于歐盟以內(nèi)的需要辦理ISO13485及CE認(rèn)證�����,MDI類(不滅菌��、非測(cè)量)可以自我申明�����,不需發(fā)證;其他非歐盟以內(nèi)的國(guó)家需要在CE認(rèn)證的基礎(chǔ)上辦理自由銷售證書(CFS)����。
IVD印度注冊(cè)需要準(zhǔn)備資料有:印度使館認(rèn)證Form9(源于法規(guī)有模板),貿(mào)促會(huì)認(rèn)證資料:自由銷售證書FSC�����,質(zhì)量管理體系類文件ISO13485�����,ISO9001,CE認(rèn)證類文件EC certificate和 自我聲明��,標(biāo)簽說明書和分析報(bào)告COA�����。
IVD敘利亞注冊(cè)需要準(zhǔn)備資料有:SDS(SAFETY DATA SHEET)����,敘利亞使館認(rèn)證資料:自由銷售證書FSC����,質(zhì)量管理體系類文件ISO13485�����,ISO9001,CE認(rèn)證類文件EC certificate和 自我聲明�����,營(yíng)業(yè)執(zhí)照��,銷售授權(quán)書����。此外需要提交說明書�����。
IVD坦桑尼亞注冊(cè)需要準(zhǔn)備資料有:中國(guó)產(chǎn)品注冊(cè)證����,其他國(guó)注冊(cè)證,質(zhì)量管理體系類文件ISO13485��,ISO9001,CE認(rèn)證類文件EC certificate和 自我聲明����,此外需要提交COA(Certificate of analysis)��。
IVD泰國(guó)注冊(cè)需要準(zhǔn)備資料有:SDS(SAFETY DATA SHEET)����,泰國(guó)使館認(rèn)證資料:自由銷售證書FSC��,質(zhì)量管理體系類文件ISO13485��,ISO9001,其他資料:銷售授權(quán)書����。此外需要提交標(biāo)簽,說明書��。
IVD柬埔寨注冊(cè)需要準(zhǔn)備資料有:貿(mào)促會(huì)認(rèn)證FSC 非認(rèn)證資料:申請(qǐng)表 (其法規(guī)有模板)��,質(zhì)量管理體系類文件ISO13485�����,ISO9001,CE認(rèn)證類文件EC certificate和 自我聲明����,標(biāo)簽,說明書和銷售授權(quán)函����,此外每個(gè)試劑還需提交兩盒樣品。
IVD黑山注冊(cè)需要準(zhǔn)備資料有:貿(mào)促會(huì)認(rèn)證資料:質(zhì)量管理體系類文件ISO13485��,ISO9001, 自由銷售證書FSC�����,產(chǎn)品保險(xiǎn)保單函��。CE認(rèn)證類文件EC certificate和 自我聲明�����,非認(rèn)證資料:標(biāo)簽�����,說明書和銷售授權(quán)函��,一套產(chǎn)品的GMDN & EDMS 碼報(bào)告��。
IVD玻利維亞注冊(cè)需要準(zhǔn)備資料有:玻利維亞使館認(rèn)證FSC 貿(mào)促會(huì)認(rèn)證資料:質(zhì)量管理體系類文件ISO13485��,ISO9001,非認(rèn)證資料CE認(rèn)證類文件EC certificate和自我聲明��,包材和標(biāo)簽����,COA(certificate of analysis)說明書和終產(chǎn)品性能評(píng)估報(bào)告,年度穩(wěn)定性報(bào)告����,銷售授權(quán)函,一套標(biāo)簽及包材樣品����。
IVD埃塞俄比亞注冊(cè)需要準(zhǔn)備資料有:埃塞俄比亞使館認(rèn)證FSC 非認(rèn)證資料:兩表格Application and Consent form (其法規(guī)有模板),質(zhì)量管理體系類文件ISO13485��,ISO9001�����,CE認(rèn)證類文件EC certificate和 自我聲明����,包材和標(biāo)簽����,說明書和性能評(píng)估報(bào)告����,穩(wěn)定性報(bào)告����,監(jiān)測(cè)和測(cè)量程序,儀器操作手冊(cè)����,儀器軟件確認(rèn)和驗(yàn)證報(bào)告,儀器性能評(píng)估報(bào)告��,儀器標(biāo)簽說明書����,他國(guó)注冊(cè)證,技術(shù)文件總結(jié)summary of technical documentstion (STED)����,基本要求檢查表EP,銷售授權(quán)函�����。
結(jié)語
不同國(guó)家或者地區(qū)對(duì)于醫(yī)療器械有不同的注冊(cè)要求,國(guó)外買家會(huì)針對(duì)性地向生產(chǎn)商提出當(dāng)?shù)氐淖?cè)要求����。
在眾多的注冊(cè)要求中,相對(duì)而言��,CE認(rèn)證及FDA注冊(cè)在全球范圍內(nèi)認(rèn)可度較高��,所以很多廠商會(huì)選擇CE認(rèn)證或者FDA注冊(cè)作為打開國(guó)際市場(chǎng)的敲門磚��,消除貿(mào)易壁壘��,增加產(chǎn)品國(guó)際影響力��,從而快速進(jìn)入國(guó)際市場(chǎng)��。