罕見病通常發(fā)病率/患病率極低��,多為慢性����、遺傳性疾病,常常危及生命��。歐美國家認(rèn)識到保障罕見病患者用藥可及性的重要性��,出臺了罕見病用藥(也稱孤兒藥)相關(guān)法律法規(guī)及制度�����,通過明確孤兒藥認(rèn)定標(biāo)準(zhǔn),給予經(jīng)過認(rèn)定的孤兒藥一系列研發(fā)����、生產(chǎn)、上市后激勵(lì)政策����,保障孤兒藥可及性��。其中����,市場獨(dú)占制度是國際通行的激勵(lì)孤兒藥研發(fā)上市的重要策略,是為了保障罕見病患者能公平地獲得健康權(quán)而采取的政策傾斜����。
獨(dú)占(exclusivity)指在藥品上市保護(hù)期,根據(jù)法規(guī)規(guī)定��,延遲或者禁止競爭藥品上市�����。與專利相比,在性質(zhì)上�����,獨(dú)占是藥品監(jiān)督管理部門提供的一種行政保護(hù)��,專利是專利行政部門在藥品開發(fā)過程中授予的財(cái)產(chǎn)權(quán)[1]在保護(hù)期限上����,獨(dú)占通常從藥品上市之日開始計(jì)算,專利可以隨時(shí)頒發(fā)或到期�����,與藥物的批準(zhǔn)狀態(tài)無關(guān)����,二者可以同時(shí)或不同時(shí)運(yùn)行[2]。獨(dú)占涉及針對創(chuàng)新藥的數(shù)據(jù)保護(hù)(數(shù)據(jù)獨(dú)占)����,以及針對孤兒藥、完成兒科研究計(jì)劃品種的市場獨(dú)占[3��,4]。理論上��,數(shù)據(jù)保護(hù)僅針對申請人自行取得��、未公開披露的試驗(yàn)數(shù)據(jù)����,并不禁止仿制藥企業(yè)自行開展臨床試驗(yàn)獲取數(shù)據(jù);市場獨(dú)占不涉及試驗(yàn)數(shù)據(jù)保護(hù)��。但在數(shù)據(jù)保護(hù)下����,仿制藥企業(yè)不會(huì)選擇自行開展臨床試驗(yàn)�����,數(shù)據(jù)保護(hù)和市場獨(dú)占往往能達(dá)到相似的市場獨(dú)占效果��。
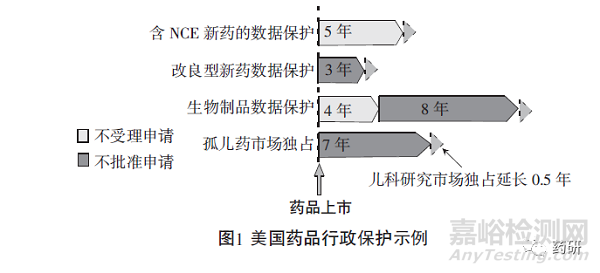
2002年�����,我國《藥品管理法實(shí)施條例》提到對獲得生產(chǎn)或者銷售含有新型化學(xué)成份藥品許可的生產(chǎn)者或者銷售者提交的自行取得且未披露的試驗(yàn)數(shù)據(jù)和其他數(shù)據(jù)實(shí)施保護(hù)�����,保護(hù)期6年,但該項(xiàng)制度一直未得到有效落實(shí)�����。2017年����,《關(guān)于深化審評審批制度改革鼓勵(lì)藥品醫(yī)療器械創(chuàng)新的意見》提出,完善和落實(shí)藥品試驗(yàn)保護(hù)����。2018年,國家藥品監(jiān)督管理局(以下簡稱國家藥監(jiān)局)就《藥品試驗(yàn)數(shù)據(jù)保護(hù)實(shí)施辦法(暫行)》公開征求意見����,提出給予罕見病治療藥品數(shù)據(jù)保護(hù)期范圍。2022年�����,《藥品管理法實(shí)施條例(征求意見稿)》首次提及對罕見病新藥給予最長不超過7年的市場獨(dú)占期����,對兒童用藥給予最長不超過12個(gè)月的市場獨(dú)占期,其間不再批準(zhǔn)相同品種上市。市場獨(dú)占制度對激勵(lì)企業(yè)開展相關(guān)領(lǐng)域藥品研發(fā)具有重要作用����。美國和歐盟的市場獨(dú)占制度運(yùn)行已有較長時(shí)間,積累了豐富的經(jīng)驗(yàn)��,本文試圖對其進(jìn)行剖析��,為我國制定罕見病用藥市場獨(dú)占制度提供參考和借鑒��。
1��、 美國孤兒藥市場獨(dú)占制度
1.1 美國孤兒藥市場獨(dú)占相關(guān)規(guī)定
為鼓勵(lì)罕見病用藥研發(fā)��,美國于1983年頒布了《孤兒藥法案》(Orphan Drug Act����,ODA)��,明確了罕見病及孤兒藥的定義����、市場獨(dú)占等內(nèi)容。1984年�����,罕見病標(biāo)準(zhǔn)修訂為“在美國患病人數(shù)不超過20萬人的疾病,或者患病人數(shù)超過20萬人但治療藥物在美國的銷售額無法補(bǔ)償其研發(fā)成本的疾病”��。用于預(yù)防�����、診斷��、治療罕見病的藥物即孤兒藥��。藥物在獲得孤兒藥資格認(rèn)定后�����,可以享受7年市場獨(dú)占����、申請費(fèi)用減免等激勵(lì)政策?���!豆聝核幏ò浮返某雠_為世界罕見病用藥政策制定奠定了基礎(chǔ)[6]。
依據(jù)《孤兒藥法案》����,獲得孤兒藥認(rèn)定的藥物在上市申請獲批時(shí)��,F(xiàn)DA若尚未批準(zhǔn)過相同藥物用于相同用途或適應(yīng)癥�����,則在上市之日起7年內(nèi)不會(huì)再批準(zhǔn)其他相同藥物用于相同用途或適應(yīng)癥�����,除非下列情形出現(xiàn):FDA撤回已上市孤兒藥的市場獨(dú)占權(quán)或撤銷其孤兒藥身份��;因任何原因撤回有關(guān)藥物的上市申請����;已上市孤兒藥持有人同意��;已上市孤兒藥持有人未能確保該藥的供應(yīng)�����。如果已有相同藥物用于相同用途或適應(yīng)癥��,后續(xù)藥物若能證明相比已上市藥物更具臨床優(yōu)勢��,則不視為“相同藥物”�����,可作為孤兒藥獲批上市并享受相關(guān)優(yōu)惠政策����。
市場獨(dú)占權(quán)是針對適應(yīng)癥的市場保護(hù),如果已存在針對該適應(yīng)癥的孤兒藥�����,F(xiàn)DA可以批準(zhǔn)該藥用于其他用途或適應(yīng)癥�����;若相同藥物的另一用途或適應(yīng)癥獲得孤兒藥資格認(rèn)定�����,F(xiàn)DA也可以授予其新的市場獨(dú)占權(quán)����;已上市藥品的新適應(yīng)癥被認(rèn)定為孤兒藥,也可獲得新的獨(dú)占期����。
1.2 孤兒藥市場獨(dú)占的關(guān)鍵要素
1.2.1 相同藥物
相同藥物主要從藥物結(jié)構(gòu)與藥物用途兩方面界定��。其中�����,小分子藥物結(jié)構(gòu)主要考慮“活性部分”(active moiety)的范圍��,常規(guī)新藥批準(zhǔn)中通常使用“活性成分”(activeingredient)的概念�����,二者有所不同����?���;钚圆糠质侵钢饕l(fā)揮生理/藥理作用的分子或離子,不包括藥物其他分子附加部分��?����;钚猿煞质侵柑峁┧幚砘钚曰蚱渌苯幼饔?�,或影響人體或其他動(dòng)物身體的結(jié)構(gòu)或任何功能的任何成分��,包括藥物生產(chǎn)過程可能經(jīng)歷化學(xué)變化并以提供指定活性或效果的改良形式存在于藥品中的組分�����。相較“活性成分”����,“活性部分”的范圍更狹窄,但依照其定義����,藥物活性成分的變化若不影響發(fā)揮生理/藥理作用的活性部分,二者會(huì)被視為相同藥物�����。在美國�����,《關(guān)于修訂<聯(lián)邦食品�����、藥品和化妝品法>的法案》授權(quán)FDA在判斷新藥是否享有獨(dú)占權(quán)等活動(dòng)中使用“活性部分”的概念。
《孤兒藥法案》頒布之初�����,僅制定了小分子藥物與大分子藥物的“相同藥物”界定標(biāo)準(zhǔn),小分子藥物在“相同藥物”界定中考慮藥物活性部分與用途兩個(gè)方面�����;大分子藥物因其結(jié)構(gòu)復(fù)雜�����,被分為蛋白質(zhì)、多糖��、多核苷酸藥物與其他四類�����,含有相同的主要分子結(jié)構(gòu)特征并用作相同用途�����,也被視為“相同藥物”��,并不要求新申請藥物含有已上市藥物的所有結(jié)構(gòu)特征,微小的結(jié)構(gòu)差異也在市場獨(dú)占的保護(hù)范圍內(nèi)�����。隨著基因技術(shù)的發(fā)展�����,2021年FDA發(fā)布了《解讀<孤兒藥法案>中的基因治療產(chǎn)品的相同性》指南,明確了用于相同適應(yīng)癥的基因治療產(chǎn)品的相同性界定原則[7]����,將轉(zhuǎn)基因與載體等某些關(guān)鍵特征視為藥物的“主要分子結(jié)構(gòu)特征”�����,同樣將藥物結(jié)構(gòu)的微小差異納入了市場獨(dú)占的保護(hù)范圍����。
1.2.2 臨床優(yōu)勢
臨床優(yōu)勢是指相較已批準(zhǔn)藥物,新申請藥物在一個(gè)或多個(gè)方面存在顯著治療優(yōu)勢����。在美國��,若能證明臨床優(yōu)勢,新申請藥物便不會(huì)被視為“相同藥物”����,可作為孤兒藥獲批上市��,并享受相應(yīng)的政策優(yōu)惠����。例如�����,2022年治療慢性髓細(xì)胞性白血?����。–ML)的孤兒藥Dasynoc被批準(zhǔn)上市�����,在其之前已有相同藥物治療相同適應(yīng)癥����,但Dasynoc在CML人群同時(shí)使用抗酸劑的情況下仍可被吸收(其他同類藥物可能需要胃酸才能被吸收)�����,符合在其他方面對患者護(hù)理作出了重大貢獻(xiàn)(Major Contribution to Patient Care����,MC toPC)標(biāo)準(zhǔn)�����,獲得了7年市場獨(dú)占期[8]����。
總的來說��,F(xiàn)DA主要從有效性、安全性對患者護(hù)理的重大貢獻(xiàn)三方面考察藥物的臨床優(yōu)勢��。(1)相比已上市的藥物具有更好的有效性�����。(2)消除與相對頻繁的不良反應(yīng)相關(guān)的成分或污染物��,提高了大部分目標(biāo)人群的安全性�����。(3)未顯示更好的有效性或安全性的情況下����,證明藥物在其他方面對患者護(hù)理作出了重大貢獻(xiàn)����。在適用于嚴(yán)重或危及生命的疾病時(shí)�����,MC to PC通?�?紤]方便的治療地點(diǎn)�����、治療持續(xù)時(shí)間、患者舒適度��、較長的給藥間隔等因素,不考慮藥物的治療成本與依從性[9]��。
2�����、 歐盟孤兒藥市場獨(dú)占制度
2.1 歐盟孤兒藥市場獨(dú)占相關(guān)規(guī)定
歐盟《孤兒藥法規(guī)》(EC No 141/2000)確定了孤兒藥認(rèn)定標(biāo)準(zhǔn)與市場獨(dú)占、費(fèi)用減免等優(yōu)惠政策��,如孤兒藥可以享受費(fèi)用減免��、市場獨(dú)占等政策激勵(lì)����。歐盟孤兒藥認(rèn)定標(biāo)準(zhǔn):用于診斷或預(yù)防嚴(yán)重威脅生命�����、慢性削弱性疾病,且患病人數(shù)不超過萬分之五�����;或沒有激勵(lì)措施的情況下��,藥物的稅后收益不足以彌補(bǔ)研發(fā)投入�����。此外,若對某適應(yīng)癥已存在相應(yīng)藥品或治療方法��,則新申請上市的孤兒藥應(yīng)證明其具有顯著收益�����。
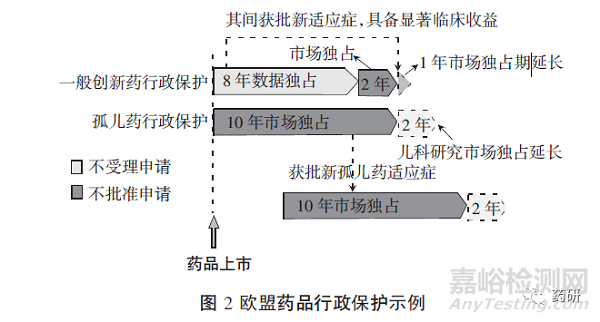
在兒科研究方面,根據(jù)法規(guī)EC No 1901/2006��,非孤兒藥完成兒科研究并符合相關(guān)規(guī)定��,或因適應(yīng)癥只影響成人等原因被豁免兒科研究��,無論該藥最終是否可以用于兒童��,都可獲得6個(gè)月的補(bǔ)充保護(hù)證書(SPC)用于延長專利�����;對孤兒藥��,完成兒科研究��,并將研究結(jié)果反映在相關(guān)文件����、包裝中或被豁免兒科研究����,可以獲得兩年的獨(dú)占期延長(見圖2)。
《孤兒藥法規(guī)》指出����,獲得孤兒藥認(rèn)定的藥物上市后��,在不損害知識產(chǎn)權(quán)法或其他歐盟法律等的情況下��,歐盟及成員國10年內(nèi)不得針對相同適應(yīng)癥的相同藥物����,接受�����、批準(zhǔn)或延長另一上市申請����,除非出現(xiàn)下列情形:原孤兒藥持有人同意;原孤兒藥持有人無法提供足夠數(shù)量的藥品����;后續(xù)申請人可以在上市申請中證明產(chǎn)品在安全性、有效性或其他方面具有臨床優(yōu)勢����。同時(shí)�����,如果在第五年底����,該孤兒藥不再符合上述認(rèn)定標(biāo)準(zhǔn)��,尤其是現(xiàn)有證據(jù)證明該孤兒藥已獲得足夠的利潤,沒有理由繼續(xù)維持市場獨(dú)占時(shí)����,其市場獨(dú)占期可縮短為6年[10]。不同于一般創(chuàng)新藥新增適應(yīng)癥并具有顯著臨床收益時(shí)增加1年市場獨(dú)占期��,孤兒藥新增適應(yīng)癥時(shí)����,新適應(yīng)癥將開啟另一個(gè)“10+2”市場獨(dú)占�����。對孤兒藥,“10+2”市場獨(dú)占與“8+2+1”的常規(guī)保護(hù)同時(shí)運(yùn)行�����,二者并行不悖[12]����。
2.2 孤兒藥市場獨(dú)占的關(guān)鍵要素
2.2.1 相同藥物
2018年,歐盟通過實(shí)施條例(EU)2018/781完善了“相同藥物”的定義����。“相同藥物”是指含有與已上市的孤兒藥中一種或多種相似活性物質(zhì)的醫(yī)藥產(chǎn)品,并用于相同的適應(yīng)癥��。其中��,“相似活性物質(zhì)”(similar active substance)可以是相同活性物質(zhì)��,也可以是具有相同主要分子結(jié)構(gòu)特征和作用機(jī)制的活性物質(zhì)��,與美國一樣�����,用途相同的藥物即便在結(jié)構(gòu)上具有微小差異����,歐盟也視為“相同藥物”。
2.2.2 臨床優(yōu)勢
與美國孤兒藥規(guī)定相近��,歐盟根據(jù)實(shí)施條例(EC)No 847/2000�����,“臨床優(yōu)勢”是指在有效性、安全性或?qū)颊咦o(hù)理的重大貢獻(xiàn)方面優(yōu)于已上市的孤兒藥��。
3����、 美國和歐盟孤兒藥市場獨(dú)占制度比較
3.1 共性及差異之處
《孤兒藥法案》與《孤兒藥法規(guī)》頒布以來��,歐美孤兒藥認(rèn)定與上市數(shù)量激增����。1983—2020年,美國認(rèn)定5 769個(gè)孤兒藥適應(yīng)癥�����,其中1 039個(gè)成功上市[12]��;2000—2020年����,歐盟對2 282個(gè)適應(yīng)癥藥物授予了孤兒藥身份,最終220個(gè)上市[13]��。《孤兒藥法案》作為世界罕見病用藥政策的基礎(chǔ)����,被視為有效衛(wèi)生政策的典范[14],市場獨(dú)占被認(rèn)為是其中最強(qiáng)有力的激勵(lì)政策�����。美國與歐盟的罕見病用藥市場獨(dú)占制度核心都是通過額外的市場利潤激勵(lì)制藥企業(yè)研發(fā)�����,并明確界定了“相同藥物”及“臨床優(yōu)勢”����。前者主要考慮藥物結(jié)構(gòu)與用途;后者主要考慮藥品安全性�����、有效性或?qū)颊咦o(hù)理的重大貢獻(xiàn)����。但二者期限不同,美國的罕見病用藥+完成兒科研究能夠獲得7+0.5年的市場獨(dú)占��,歐盟可以獲得10+2年;同時(shí)�����,歐盟在第五年底設(shè)置了評估流程�����,試圖將獲得足夠利潤的罕見病用藥市場獨(dú)占期收縮至6年��。
3.2 存在的問題
3.2.1 相同適應(yīng)癥的界定存在爭議
盡管歐美國家已圍繞罕見病用藥市場獨(dú)占制定并完善了“相同藥物”“臨床優(yōu)勢”等關(guān)鍵要素的界定標(biāo)準(zhǔn)��,但仍有新的爭議出現(xiàn)�����,例如對“相同適應(yīng)癥”覆蓋范圍的爭議��。美國巡回法院將其解讀為疾病整體����,但目前仍存在爭議�����,《孤兒藥法案》可能會(huì)進(jìn)一步修訂,將市場獨(dú)占限制在“經(jīng)批準(zhǔn)的用途或適應(yīng)癥”范圍內(nèi)����。
3.2.2 市場獨(dú)占保護(hù)作用減弱
市場獨(dú)占制度制定之初,為難以獲得專利保護(hù)的生物制品提供了有力的行政保護(hù)[15]����,但隨著專利制度的完善與生物制品數(shù)據(jù)保護(hù)制度的頒布,市場獨(dú)占的保護(hù)作用相對減弱����。有研究指出,1985—1994年��,美國僅有57%的孤兒藥擁有專利�����,50%的孤兒藥市場獨(dú)占期超過專利期��;1995—2004年��,這兩個(gè)數(shù)字變?yōu)?7%與35%�����;到2005—2014年,87%的孤兒藥擁有專利�����,僅18%的孤兒藥市場獨(dú)占期超過專利期����。可見����,專利對孤兒藥的保護(hù)越來越全面,市場獨(dú)占期超過專利期的孤兒藥比例越來越低��,對孤兒藥市場競爭的保護(hù)作用越來越弱[15]��。
3.2.3 定期評估條款無法發(fā)揮作用
孤兒藥認(rèn)定是針對藥物特定適應(yīng)癥的資格認(rèn)定����,同一藥物可通過不同適應(yīng)癥獲得多個(gè)孤兒藥認(rèn)定與市場獨(dú)占����,這種模式使一種藥物獲取超額利潤成為可能。對此����,歐盟試圖以定期評估的方式平衡企業(yè)利益和公眾利益�����,但收效甚微��。一是雖然法規(guī)指出在第5年底對孤兒藥進(jìn)行評估�����,但實(shí)際僅評估通過“收入不足以彌補(bǔ)研發(fā)成本”這一路徑獲得認(rèn)定的孤兒藥��,而非將所有孤兒藥納入評估范圍�����。二是“足夠的利潤”這一概念未有明確的界定[16]�����。
4����、 啟示
我國于2022年頒布《藥品管理法實(shí)施條例(征求意見稿)》(以下簡稱《條例》),首次提及市場獨(dú)占期:對首個(gè)批準(zhǔn)上市的兒童專用新品種�����、劑型和規(guī)格��,及增加兒童適應(yīng)癥或用法用量的�����,給予最長不超過12個(gè)月的市場獨(dú)占期����;對批準(zhǔn)上市的罕見病新藥,在藥品上市許可有人承諾保障藥品供應(yīng)情況下����,給予最長不超過7年的市場獨(dú)占期,期間不再批準(zhǔn)相同品種上市����,藥品上市許可持有人不履行供應(yīng)保障承諾的��,終止市場獨(dú)占期�����。對此,結(jié)合歐美國家市場獨(dú)占制度規(guī)定及其實(shí)施經(jīng)驗(yàn)����,本文提出以下思考與建議。
4.1 明確罕見病用藥市場獨(dú)占適用范圍
《條例》將罕見病用藥的市場獨(dú)占保護(hù)范圍限制在“罕見病新藥”����,根據(jù)2015年《國務(wù)院關(guān)于改革藥品醫(yī)療器械審評審批制度的意見》,“新藥”指“未在中國境內(nèi)外上市銷售的藥品”��,即“全球新”�����;根據(jù)物質(zhì)基礎(chǔ)的原創(chuàng)性和新穎性�����,又分為創(chuàng)新藥和改良型新藥[17]����。在歐美國家,孤兒藥認(rèn)定資格并未與藥品注冊分類“綁定”�����,更強(qiáng)調(diào)在本國市場中尚未有相同藥物用于某適應(yīng)癥。目前����,我國孤兒藥自主研制市場幾近空白,主要依賴進(jìn)口[18]�����。罕見病用藥市場獨(dú)占的適用范圍若依照“全球新”��,一是對引入國外已上市藥物無法起到激勵(lì)作用�����;二是專利已過期的“老藥”若無行政保護(hù)作為補(bǔ)償�����,不會(huì)選擇進(jìn)入中國市場�����;三是“新藥”的范圍無法鼓勵(lì)我國企業(yè)對已過專利期的藥物進(jìn)行仿制��。因此����,需結(jié)合我國罕見病用藥國情,明確罕見病用藥市場獨(dú)占的適用范圍�����,盡快提升罕見病用藥的可及性��。
4.2 明晰兒科用藥市場獨(dú)占性質(zhì)
罕見病用藥的市場獨(dú)占是一種初始獨(dú)占��,即從藥品上市之日起計(jì)算����;兒科用藥的市場獨(dú)占時(shí)間較短,是一種附加獨(dú)占��,用于延長其他市場獨(dú)占或?qū)@?���,且不要求研究結(jié)果為陽性,即只要符合相關(guān)兒科研究計(jì)劃��,即使最終該藥不可用于兒童�����,也可以享有兒科用藥市場獨(dú)占。我國《條例》要求藥品可以用于兒童�����,對企業(yè)而言����,開展兒科臨床研究的風(fēng)險(xiǎn)更大,但最高12個(gè)月的兒童用藥市場獨(dú)占性質(zhì)不明確����,若與專利或其他市場獨(dú)占并行,則激勵(lì)效果有限����。罕見病大多發(fā)病于兒童期[19],我國應(yīng)鼓勵(lì)罕見病用藥兒科研究����,以滿足兒童患者的臨床需求。在兩種市場獨(dú)占并行的情況下����,兒童用藥市場獨(dú)占失去了意義�����,尤其是當(dāng)開展兒科用藥研究成本高、風(fēng)險(xiǎn)高的情形下�����,在成人用藥基礎(chǔ)上進(jìn)行超說明書用藥����,可能對制藥企業(yè)更有利。因此�����,應(yīng)明確兒童用藥市場獨(dú)期能否與罕見病用藥市場獨(dú)占期“串聯(lián)”運(yùn)作��。
4.3 確定市場獨(dú)占中關(guān)鍵要素的概念及界定標(biāo)準(zhǔn)
第一�����,明確“相同品種”的定義�����。《條例》指出����,市場獨(dú)占期期間,不再批準(zhǔn)“相同品種”上市�����,但需進(jìn)一步明確“相同品種”這一概念����。例如美國以分子的“活性部分”界定小分子藥物市場獨(dú)占的“相同藥物”,替換了“活性成分”概念��,靈活地收縮了界定范圍����,擴(kuò)大了孤兒藥市場獨(dú)占的保護(hù)范圍;同時(shí)歐美都以法規(guī)中的定義部分或出臺條例的形式對“相同藥物”作了詳細(xì)規(guī)定��。
第二��,充分考慮“相同適應(yīng)癥”要素����。無論是市場獨(dú)占制度的核心內(nèi)容��,還是“相同藥物”的界定�����,歐美國家均強(qiáng)調(diào)“適應(yīng)癥”這一要素��。市場獨(dú)占制度的本質(zhì)是在獨(dú)占期期間,給予特定適應(yīng)癥的市場保護(hù)�����。相同品種藥品用于其他適應(yīng)癥的上市申請��,乃至另一市場獨(dú)占權(quán)的授予����,都不應(yīng)受已上市品種市場獨(dú)占期的限制。
第三����,制定“臨床優(yōu)勢”相關(guān)標(biāo)準(zhǔn)。罕見病用藥激勵(lì)政策的初衷是通過額外的經(jīng)濟(jì)利潤激勵(lì)制藥企業(yè)研發(fā)��,提高罕見病用藥可及性。因此�����,當(dāng)新申請藥物滿足“相同品種”界定標(biāo)準(zhǔn)時(shí)��,不應(yīng)直接因市場獨(dú)占而拒絕批準(zhǔn)其上市申請����,而是應(yīng)由后續(xù)申請人自證其藥品相比已批準(zhǔn)藥品更具臨床優(yōu)勢。“臨床優(yōu)勢”概念可借鑒歐美國家的成熟經(jīng)驗(yàn)��,考慮藥品安全性��、有效性或?qū)颊咦o(hù)理的重大貢獻(xiàn)��,同時(shí)對相關(guān)數(shù)據(jù)的獲取方式作出要求��。