近日�����,上海器審中心發(fā)布《增材制造定制式骨科手術(shù)導(dǎo)板注冊審評指南》����,內(nèi)容如下:
本審評指南旨在指導(dǎo)注冊申請人對增材制造定制式骨科手術(shù)導(dǎo)板注冊申報資料的準(zhǔn)備及撰寫,同時也為技術(shù)審評部門提供參考��。
本審評指南是對增材制造定制式骨科手術(shù)導(dǎo)板的一般要求�����,注冊申請人應(yīng)依據(jù)產(chǎn)品的具體特性確定其中內(nèi)容是否適用���。若不適用,需具體闡述理由及相應(yīng)的科學(xué)依據(jù)��,并依據(jù)產(chǎn)品的具體特性對注冊申報資料的內(nèi)容進行充實和細化��。
本審評指南是供注冊申請人和技術(shù)審評人員使用的指導(dǎo)性文件,但不包括審評審批所涉及的行政事項���,亦不作為法規(guī)強制執(zhí)行���,應(yīng)在遵循相關(guān)法規(guī)的前提下使用本審評指南。
如果有能夠滿足相關(guān)法規(guī)要求的其他方法���,也可以采用�,但是需要提供詳細的研究資料和驗證資料�����。
本審評指南是在現(xiàn)行法規(guī)和標(biāo)準(zhǔn)體系以及當(dāng)前認知水平下制定�,隨著法規(guī)和標(biāo)準(zhǔn)的不斷完善,以及科學(xué)技術(shù)的不斷發(fā)展�,相關(guān)內(nèi)容也將適時進行調(diào)整。
一�����、適用范圍
本指南適用于第二類增材制造定制式骨科手術(shù)導(dǎo)板�����。該類產(chǎn)品為骨科手術(shù)配套手術(shù)工具����,根據(jù)電子計算機斷層掃描(CT)和磁共振成像(MRI)2 種非接觸式的獲取方法得到的數(shù)據(jù)進行三維重建后通過增材制造方式制造而成,在使用過程中不會接觸中樞神經(jīng)系統(tǒng)或血液循環(huán)系統(tǒng)����,一般采用具有良好生物安全性的高分子材料。用于骨科手術(shù)中定位��、探測����、導(dǎo)向、評估或提供基準(zhǔn)用�。
本指南所稱增材制造工藝僅限于選擇性激光燒結(jié)(SLS)工藝且使用高分子材料加工而成。不包括使用金屬材料�、陶瓷材料或其他材料通過其它增材制造工藝加工的產(chǎn)品,但可參考本指南中適用的部分����。
常見產(chǎn)品舉例如下:脊柱手術(shù)導(dǎo)板、膝關(guān)節(jié)手術(shù)導(dǎo)板�、醫(yī)用個性化骨科手術(shù)導(dǎo)板等。
二��、注冊申報資料要求
(一)監(jiān)管信息
1.申請表
注冊申請人應(yīng)按照填表要求填寫。注冊申請人應(yīng)至少明確產(chǎn)品名稱���、分類代碼等信息�。如產(chǎn)品型號規(guī)格較多����,應(yīng)以列表形式提供,并附頁于申請表后��。
1.1 產(chǎn)品名稱的要求
增材制造定制式骨科手術(shù)導(dǎo)板的名稱可按作用對象和預(yù)期用途等方式來命名�����,如醫(yī)用骨科手術(shù)導(dǎo)板����、髖關(guān)節(jié)置換個性化手術(shù)導(dǎo)板、增材制造膝關(guān)節(jié)手術(shù)導(dǎo)板等�����。
1.2 產(chǎn)品分類信息
依據(jù)《醫(yī)療器械分類目錄》及《2018 年醫(yī)療器械產(chǎn)品分類界定結(jié)果匯總》���,增材制造定制式骨科手術(shù)導(dǎo)板管理類別為 II 類��,子目錄為 04 骨科手術(shù)器械 16 關(guān)節(jié)外科輔助器械�。注冊申請人應(yīng)根據(jù)申報產(chǎn)品的實際情況���,明確其子目錄信息���、一級產(chǎn)品類別信息及二級產(chǎn)品類別信息。
1.3 產(chǎn)品注冊單元的劃分
注冊單元劃分應(yīng)根據(jù)相關(guān)法規(guī)文件要求�,醫(yī)療器械注冊單元原則上以產(chǎn)品的技術(shù)原理、結(jié)構(gòu)組成�����、性能指標(biāo)和適用范圍為劃分依據(jù)�。
原材料不一致而導(dǎo)致產(chǎn)品性能指標(biāo)不同時建議按照不同注冊單元進行劃分。以無菌形式和非無菌形式提供的增材制造定制式骨科手術(shù)導(dǎo)板可劃為同一注冊單元����。例如醫(yī)用個性化骨科手術(shù)導(dǎo)板無菌型和非無菌型可劃為同一注冊單元。
2.產(chǎn)品列表
以表格形式列出擬申報產(chǎn)品的型號�����、規(guī)格�����、結(jié)構(gòu)及組成、附件等���,以及每個型號規(guī)格的標(biāo)識(如型號或部件的編號����,器械唯一標(biāo)識等)和描述說明(如尺寸���、材質(zhì)等)�。
(二)綜述資料
1.概述
1.1 增材制造定制式骨科手術(shù)導(dǎo)板的管理類別為 II 類醫(yī)療器械����,注冊申請人應(yīng)根據(jù)申報產(chǎn)品的實際情況,明確其子目錄信息��、一級產(chǎn)品類別信息及二級產(chǎn)品類別信息���。
1.2 產(chǎn)品名稱:增材制造定制式骨科手術(shù)導(dǎo)板的命名應(yīng)符合相關(guān)法規(guī)�、國家標(biāo)準(zhǔn)���、行業(yè)標(biāo)準(zhǔn)的要求���。通常由 1 個核心詞和不超過 3 個特征詞確定產(chǎn)品通用名稱����,并適度考慮臨床習(xí)慣�����?���?砂醋饔脤ο?����、預(yù)期用途和加工工藝等方式來命名��,一般不使用“3D 打印”��。如醫(yī)用骨科手術(shù)導(dǎo)板���、髖關(guān)節(jié)置換個性化手術(shù)導(dǎo)板����、增材制造膝關(guān)節(jié)手術(shù)導(dǎo)板。
2.器械組成��、功能及作用原理
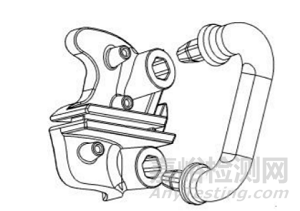
2.1 提供產(chǎn)品各型號的幾何結(jié)構(gòu)外形圖����,如圖 1 所示,充分描述產(chǎn)品結(jié)構(gòu)組成信息����。
圖 1 脛骨近端增材制造骨科導(dǎo)板結(jié)構(gòu)外形圖
2.2 申報資料中需明確增材制造用粉末材料的通用名稱、商品名�����、牌號�、CAS 號、原材料生產(chǎn)供應(yīng)商及符合的標(biāo)準(zhǔn)�����。并描述粉末材料的性能參數(shù)����,如粉末的基本形貌、粉末的化學(xué)成分、粉末純度����、粉末粒徑及粒徑分布、粉末球形度等信息��。進口材料的通用名稱�、商品名、牌號及符合標(biāo)準(zhǔn)不應(yīng)超過原產(chǎn)國上市證明文件及說明書批準(zhǔn)的范圍�����。
2.3 應(yīng)提供增材制造打印設(shè)備的性能參數(shù)����,并闡述打印設(shè)備與所選擇的粉末原材料的匹配性����。
2.4 應(yīng)提供經(jīng)過設(shè)計開發(fā)驗證確認活動后得到的增材制造工藝參數(shù),并闡述工藝機理�。
2.5 說明產(chǎn)品的型號規(guī)格及劃分依據(jù),明確各型號規(guī)格的區(qū)別����,可列表對不同型號規(guī)格的結(jié)構(gòu)組成、尺寸、性能指標(biāo)等加以描述�,也可采用示意圖進行表述。
2.6 提供產(chǎn)品工作原理/作用機理�。
2.7 提供產(chǎn)品的包裝信息,可包括包裝形式��、包裝材料����、包裝工藝、包裝原材料供應(yīng)商信息以及無菌屏障系統(tǒng)(如適用)等信息�。
2.8 產(chǎn)品的適用范圍、適用人群�、禁忌證需與申報產(chǎn)品的性能、功能相符���。適用范圍的描述需清晰準(zhǔn)確�,如描述為:作為踝上截骨手術(shù)的輔助工具���,輔助術(shù)者進行截骨和矯正下肢力線��。
注冊申請人需根據(jù)申報產(chǎn)品的設(shè)計特征����,進一步說明其具體的適用人群、預(yù)期使用環(huán)境等信息����。描述產(chǎn)品的禁忌證(包括絕對禁忌證、相對禁忌證)���,如不適宜使用的人群��、疾病等情形�。
2.9 提供同類產(chǎn)品(國內(nèi)外已上市)或前代產(chǎn)品(如有)的信息�,闡述申請注冊產(chǎn)品的研發(fā)背景和目的。對于同類產(chǎn)品�����,說明選擇其作為研發(fā)參考的原因�。
注冊申請人需列表比較說明本次申報產(chǎn)品與已上市同類或前代產(chǎn)品(如有)的相同點和不同點����,比較的項目包括產(chǎn)品名稱、工作原理���、原材料��、結(jié)構(gòu)特點�、性能指標(biāo)、適用范圍���、生產(chǎn)工藝����、滅菌方式(如適用)�、有效期,以及與市場上同類產(chǎn)品在技術(shù)����、設(shè)計和應(yīng)用方面的比較資料等。
(三)非臨床資料
1.產(chǎn)品風(fēng)險管理資料
注冊申請人需對產(chǎn)品全生命周期實施風(fēng)險管理�,應(yīng)參照GB/T 42062-2022《醫(yī)療器械 風(fēng)險管理對醫(yī)療器械的應(yīng)用》(2022.10.12 發(fā)布,2023.11.01 實施)提交風(fēng)險管理資料��。應(yīng)對該類產(chǎn)品進行充分的風(fēng)險識別����,風(fēng)險識別的信息來源需要具體列出,可包括但不局限于以下途徑:類似產(chǎn)品的投訴/抱怨數(shù)據(jù)����、醫(yī)學(xué)文獻��、實驗室檢測����、產(chǎn)品標(biāo)簽標(biāo)識等����。對于風(fēng)險識別信息的來源注冊申請人應(yīng)具體說明,并提交有關(guān)支持文
件或文獻����。
注冊申請人在產(chǎn)品注冊上市前,需對風(fēng)險管理過程進行評審�����。評審需至少確保:風(fēng)險管理計劃已被適當(dāng)?shù)貙嵤?�,綜合剩余風(fēng)險是可接受的�。評審結(jié)果需形成風(fēng)險管理報告�。申報資料格式需符合現(xiàn)行有效的法規(guī)文件的要求。
除無源醫(yī)療器械已識別的共性風(fēng)險外��,對于增材制造定制式骨科手術(shù)導(dǎo)板類產(chǎn)品��,注冊申請人至少還需關(guān)注以下方面的風(fēng)險:
1.1 生產(chǎn)加工過程可能產(chǎn)生的危害:
污染;
添加劑(助劑)的殘留��;
生產(chǎn)工藝參數(shù)的改變���;如激光功率����、掃描速率改變等��;
打印過程中生產(chǎn)人員吸入粉末����;
打印過程中惰性保護氣體的泄漏。
1.2 物理機械性能
與使用者接觸的部分�,存在銳邊、毛刺等���。
1.3 產(chǎn)品說明書及標(biāo)簽
說明書中沒有提供相關(guān)信息���,如清洗、滅菌方法��、使用前的檢查建議等���;
說明書警示信息不明確���;
對副作用警告不充分�����;
使用操作說明不規(guī)范����。
2.產(chǎn)品技術(shù)要求
注冊申請人應(yīng)結(jié)合產(chǎn)品的技術(shù)特征和臨床使用情況來編制技術(shù)要求����,對宣稱的所有技術(shù)參數(shù)和功能,應(yīng)在產(chǎn)品技術(shù)要求中予以規(guī)定�����;產(chǎn)品技術(shù)要求中的內(nèi)容引用國家標(biāo)準(zhǔn)����、行業(yè)標(biāo)準(zhǔn)的應(yīng)保證其適用性。若以下相關(guān)性能指標(biāo)要求(包括國家標(biāo)準(zhǔn)或行業(yè)標(biāo)準(zhǔn)中規(guī)定的要求)未適用�����,注冊申請人應(yīng)在提交注冊材料的研究資料中對未適用情況進行合理的說明���。
產(chǎn)品技術(shù)要求應(yīng)包括但不局限于以下內(nèi)容:
2.1 表面質(zhì)量
2.1.1 外觀
外管整潔��,表面無鋒棱���、毛刺和裂紋等缺陷,無殘存支撐材料�、粉末碎屑等附著物。
增材制造定制式骨科手術(shù)導(dǎo)板與骨骼模型進行貼合��,間隙不大于 1mm�。
增材制造定制式骨科手術(shù)導(dǎo)板與骨骼模型進行貼合,貼合位置唯一����。
2.1.2 表面粗糙度
應(yīng)符合規(guī)定產(chǎn)品表面粗糙度的要求。如有國家標(biāo)準(zhǔn)���、行業(yè)標(biāo)準(zhǔn)���,應(yīng)按相應(yīng)標(biāo)準(zhǔn)執(zhí)行且應(yīng)滿足臨床使用要求。
2.2 尺寸
應(yīng)明確產(chǎn)品規(guī)格尺寸和公差,尺寸允許公差參照應(yīng)參照行業(yè)標(biāo)準(zhǔn)中(YY/T 0940��、YY/T 0941�����、YY/T 0943����、YY/T 0944等標(biāo)準(zhǔn))的相關(guān)要求。若注冊申請人自行設(shè)定�����,應(yīng)提供公差設(shè)定的依據(jù)����。
尺寸應(yīng)符合產(chǎn)品設(shè)計規(guī)范的要求和臨床醫(yī)生提出的具體制備需求,在確定導(dǎo)板設(shè)計方案�����、手術(shù)入路�、顯露范圍、安放位置��、貼附區(qū)域后方可制作相應(yīng)尺寸大小的導(dǎo)板。
2.3 力學(xué)性能
注:由于尺寸�、形狀等因素?zé)o法取樣測試時,可使用相同材料和制造工藝的試件進行測試��。
2.3.1 拉伸斷裂強度�、斷裂伸長率����、彎曲強度應(yīng)規(guī)定導(dǎo)板的拉伸斷裂強度、斷裂伸長率�、彎曲強度的要求。如有國家標(biāo)準(zhǔn)���、行業(yè)標(biāo)準(zhǔn)要求�����,應(yīng)執(zhí)行相應(yīng)標(biāo)準(zhǔn)且應(yīng)滿足臨床使用要求���。
2.3.2 彈性模量�����、彎曲模量(如適用)
應(yīng)規(guī)定導(dǎo)板的彈性模量、彎曲模量的要求����。如有國家標(biāo)準(zhǔn)、行業(yè)標(biāo)準(zhǔn)要求����,應(yīng)執(zhí)行相應(yīng)標(biāo)準(zhǔn)且應(yīng)滿足臨床使用要求。
2.3.3 硬度
應(yīng)規(guī)定導(dǎo)板表面硬度的要求�。如有國家標(biāo)準(zhǔn)、行業(yè)標(biāo)準(zhǔn)要求����,應(yīng)執(zhí)行相應(yīng)標(biāo)準(zhǔn)且應(yīng)滿足臨床使用要求。
例如聚十二內(nèi)酰胺材料采用選擇性激光燒結(jié)(SLS)工藝制備而成的產(chǎn)品����,其硬度應(yīng)達到≥70 HRR。
2.3.4 抗摩擦性能
應(yīng)規(guī)定導(dǎo)板抗摩擦性能的要求�。如有國家標(biāo)準(zhǔn)、行業(yè)標(biāo)準(zhǔn)要求�����,應(yīng)執(zhí)行相應(yīng)標(biāo)準(zhǔn)且應(yīng)滿足臨床使用要求���。
2.3.5 沖擊強度(如適用)
應(yīng)規(guī)定導(dǎo)板沖擊強度的要求�����。如有國家標(biāo)準(zhǔn)���、行業(yè)標(biāo)準(zhǔn)要求���,應(yīng)執(zhí)行相應(yīng)標(biāo)準(zhǔn)且應(yīng)滿足臨床使用要求�����。
2.4 使用性能
導(dǎo)板相關(guān)配套零件通過應(yīng)順暢�,不得有明顯的卡塞現(xiàn)象����。零件之間裝配��、拆分等應(yīng)靈活�����、無松動、變形和斷裂現(xiàn)象����。增材制造定制式骨科手術(shù)導(dǎo)板的使用性能如有國家標(biāo)準(zhǔn)、行業(yè)標(biāo)準(zhǔn)要求���,應(yīng)執(zhí)行相應(yīng)標(biāo)準(zhǔn)且應(yīng)滿足臨床使用要求和解剖及生物力學(xué)的要求、滿足產(chǎn)品設(shè)計需求�、符合器械的安裝及使用穩(wěn)定性。
2.5 化學(xué)性能
對于高分子材料通過增材制造方式制造的定制式骨科手術(shù)導(dǎo)板����,其化學(xué)性能應(yīng)符合相應(yīng)的國家、行業(yè)標(biāo)準(zhǔn)的要求����。至少包括酸堿度、還原物質(zhì)�、重金屬含量、紫外吸光度����、蒸發(fā)殘渣等�����。但對于首次應(yīng)用于醫(yī)療器械的新材料����,應(yīng)根據(jù)該材料適合用于人體的預(yù)期使用部位�、預(yù)期使用方式等情況,
增加除以上化學(xué)性能外的其它化學(xué)要求���。
2.6 微生物限度(如適用)
若增材制造定制式骨科手術(shù)導(dǎo)板以微生物限度控制狀態(tài)提供��,按照《中華人民共和國藥典》 四部通則 非無菌產(chǎn)品微生物限度檢查:微生物計數(shù)法和非無菌產(chǎn)品微生物限度檢查:控制菌檢查法進行檢測����,結(jié)果需符合相關(guān)要求。
2.7 無菌(如適用)
增材制造定制式骨科手術(shù)導(dǎo)板經(jīng)確認的方法滅菌后應(yīng)無菌����。
2.8 環(huán)氧乙烷殘留量(如適用)
若為無菌產(chǎn)品且經(jīng)環(huán)氧乙烷滅菌,環(huán)氧乙烷殘留量應(yīng)不大于 10μg/g���。
2.9 其他
增材制造定制式骨科手術(shù)導(dǎo)板應(yīng)根據(jù)自身特性制定專有技術(shù)指標(biāo)�,應(yīng)能滿足其使用要求。
3.研究資料
3.1 產(chǎn)品性能研究
應(yīng)當(dāng)提供產(chǎn)品性能研究資料以及產(chǎn)品技術(shù)要求的研究和編制說明���,包括功能性�、安全性指標(biāo)的確定依據(jù)�,所采用的標(biāo)準(zhǔn)或方法、采用的原因及理論基礎(chǔ)等�����。此外�����,研究資料還應(yīng)重點闡述以下內(nèi)容:
3.1.1 粉末的控制研究
應(yīng)提供粉末原材料的選擇依據(jù)����,粉末原材料的物理化學(xué)性能分析,重點應(yīng)闡述與打印設(shè)備的匹配性研究���。
應(yīng)提供粉末原材料的貯存要求分析說明����,是否可回收再利用�、粉末回收次數(shù)等的確定依據(jù)及所開展的研究�����,提供相應(yīng)的物理�、化學(xué)等驗證����。
應(yīng)提供工藝流程及控制措施,如氣體保護和污染防控���、防爆控制�����。
3.1.2 打印工藝參數(shù)的控制研究
應(yīng)明確打印艙室的環(huán)境要求,闡述理由�、依據(jù)及開展的研究,闡述如何實現(xiàn)控制���。
應(yīng)針對打印工藝參數(shù)進行驗證����。如存在不同粉末���、不同打印設(shè)備匹配組合的情況����,應(yīng)結(jié)合實際詳細闡述不同匹配組合下的差異性及針對差異所開展的研究。
3.1.3 軟件���、設(shè)備的控制研究
應(yīng)提供三維圖像重建處理軟件的確認資料��、動物離體骨三維圖像重建質(zhì)量驗證資料���;應(yīng)提供激光粉末打印機設(shè)備安裝確認資料、增材制造工藝過程確認資料���、軟件確認資料等��。
3.1.4 醫(yī)工交互的控制研究
應(yīng)提供醫(yī)工交互過程控制研究資料����,如醫(yī)工交互過程控制文件及具體案例等�����。
3.2 生物相容性的評價研究
應(yīng)描述增材制造定制式骨科手術(shù)導(dǎo)板與人體接觸部件的材料,以及接觸的性質(zhì)和時間�,參照《關(guān)于印發(fā)醫(yī)療器械生物學(xué)評價和審評指南的通知》、GB/T 16886.1《醫(yī)療器械生物學(xué)評價 第 1 部分:風(fēng)險管理過程中的評價與試驗》的要求對其進行生物相容性評價證明其安全性�����。生物相容性試驗應(yīng)按照 GB/T 16886 系列標(biāo)準(zhǔn)的要求開展���。應(yīng)至少進行細胞毒性����、致敏反應(yīng)����、刺激或皮內(nèi)反應(yīng)、材料介導(dǎo)的致熱性(無菌產(chǎn)品適用�、非無菌產(chǎn)品建議滅菌后進行研究)、急性全身毒性���。
3.3 產(chǎn)品滅菌或清洗、消毒工藝研究
增材制造定制式骨科手術(shù)導(dǎo)板可根據(jù)市場需求����,以無菌形式或非無菌形式提供。注冊申請人應(yīng)對以無菌形式提供的產(chǎn)品明確其滅菌工藝(方法和參數(shù))和無菌保證水平(SAL),并提供滅菌確認報告����。如滅菌使用的方法容易出現(xiàn)殘留,應(yīng)當(dāng)明確殘留物信息及采取的處理方法�,并提供研究資料。
若產(chǎn)品使用前由醫(yī)療機構(gòu)進行滅菌�����,注冊申請人應(yīng)明確推薦的滅菌工藝(方法和參數(shù))及所推薦的滅菌方法確定的依據(jù)����;應(yīng)當(dāng)提供產(chǎn)品相關(guān)推薦的滅菌方法耐受性的研究資料。
3.4 穩(wěn)定性研究
應(yīng)明確增材制造定制式骨科手術(shù)導(dǎo)板的貨架有效期�����,并提供理論依據(jù)��。
應(yīng)明確增材制造定制式骨科手術(shù)導(dǎo)板的包裝形式并確保包裝在宣稱的運輸儲存條件下���,能夠?qū)Ξa(chǎn)品起到防護作用并保持產(chǎn)品清潔����。
應(yīng)提交運輸穩(wěn)定性驗證資料,如產(chǎn)品包裝的跌落試驗���、振蕩試驗等��,證明在規(guī)定的運輸條件下不會對產(chǎn)品的性能造成不利影響����。
因定制式產(chǎn)品的特殊性�����,應(yīng)考慮增材制造定制式骨科手術(shù)導(dǎo)板從模型獲取到術(shù)中使用期間人體骨骼形態(tài)是否會發(fā)生變化等���。
4.產(chǎn)品檢驗報告
注冊申請人應(yīng)提供產(chǎn)品檢驗報告�����,產(chǎn)品檢驗報告應(yīng)符合國務(wù)院藥品監(jiān)督管理部門的要求�����,可以是醫(yī)療器械注冊申請人的自檢報告���,也可以是委托具有醫(yī)療器械檢驗資質(zhì)的醫(yī)療器械檢驗機構(gòu)出具的檢驗報告。
注冊申請人應(yīng)提供典型性檢驗樣品的選擇說明���,所檢驗型號產(chǎn)品應(yīng)當(dāng)是本注冊單元內(nèi)能夠代表申報的其他型號產(chǎn)品安全性和有效性的典型產(chǎn)品��。若一個型號規(guī)格不能覆蓋���,應(yīng)選擇不同型號規(guī)格進行差異性檢驗。如申報產(chǎn)品型號規(guī)格包括骨盆�、膝關(guān)節(jié)、脛骨近端��、踝關(guān)節(jié)���、下肢骨干��、下頜骨�����、頸椎�、胸腰椎等部位����,不同型號的組件應(yīng)分別選取典型性產(chǎn)品進行檢驗����,應(yīng)涵蓋結(jié)構(gòu)最復(fù)雜規(guī)格�����。
(四)臨床評價資料
3D 打印截骨導(dǎo)板已列入《國家藥監(jiān)局關(guān)于發(fā)布免于進行臨床評價醫(yī)療器械目錄的通告》(以下簡稱《目錄》���,2021年第 71 號)中的免于進行臨床試驗醫(yī)療器械目錄���。序號 237,分類編碼 04-16-03�����。注冊申請人應(yīng)提交申報產(chǎn)品相關(guān)信息與”《目錄》所述內(nèi)容的對比資料���。并按《列入免于進行臨床評價醫(yī)療器械目錄產(chǎn)品對比說明技術(shù)指導(dǎo)原則》的要求提交“申報產(chǎn)品與目錄中已獲準(zhǔn)境內(nèi)注冊醫(yī)療器械對比表”��。
未列入《目錄》的增材制造骨科手術(shù)導(dǎo)板�����,注冊申請人可參照《醫(yī)療器械臨床評價技術(shù)指導(dǎo)原則》的要求�,通過同品種醫(yī)療器械臨床試驗或臨床使用獲得的數(shù)據(jù)進行分析評價,重點關(guān)注粉末原材料���、生產(chǎn)制造工藝、結(jié)構(gòu)��、作用部位等方面�����,應(yīng)當(dāng)描述申報產(chǎn)品與同品種醫(yī)療器械的差異���,提交充分的科學(xué)證據(jù)證明二者具有相同的安全有效性���;或按照相關(guān)要求開展臨床試驗進行評價。
如果選擇進行臨床試驗��,則應(yīng)嚴(yán)格按照《醫(yī)療器械臨床試驗質(zhì)量管理規(guī)范》的要求進行臨床試驗�,并提交完整的臨床試驗資料。
(五)產(chǎn)品說明書和標(biāo)簽樣稿
產(chǎn)品說明書和標(biāo)簽的編寫要求應(yīng)符合相關(guān)法規(guī)文件和相關(guān)行業(yè)標(biāo)準(zhǔn)(如:YY/T 0466.1)的要求����。所提交的文本和標(biāo)簽樣稿應(yīng)內(nèi)容清晰、完整�����。說明書中的適用范圍、禁忌證�����、注意事項��、警示信息��、有效期等信息應(yīng)與產(chǎn)品綜述資料�����、研究資料和臨床評價資料中所描述及驗證的內(nèi)容一致����。產(chǎn)品說明書還應(yīng)包括以下內(nèi)容:
1.應(yīng)明確非滅菌提供產(chǎn)品使用前的清洗、消毒或滅菌方式�;
2.應(yīng)明確滅菌提供產(chǎn)品的滅菌方式及滅菌有效期(若適用);
3.應(yīng)注明產(chǎn)品貯存�、運輸過程中的環(huán)境要求;
4.應(yīng)明確配套���、組合產(chǎn)品使用方法��;
5.應(yīng)按照相應(yīng)行業(yè)標(biāo)準(zhǔn)�����,明確增材制造定制式骨科手術(shù)導(dǎo)板的標(biāo)志�����、標(biāo)識��;
6.應(yīng)附產(chǎn)品結(jié)構(gòu)示意圖及術(shù)中示意圖����;
7.在未完整閱讀適用說明書之前請勿嘗試操作該產(chǎn)品�,任何的不謹慎操作都將給手術(shù)帶來風(fēng)險。
(六)質(zhì)量管理體系文件
注冊申請人應(yīng)當(dāng)形成相關(guān)質(zhì)量管理體系文件和記錄���。應(yīng)當(dāng)提交下列資料���,在質(zhì)量管理體系核查時進行檢查。
1.注冊申請人基本情況表�����。
2.注冊申請人組織機構(gòu)圖。
3.生產(chǎn)企業(yè)總平面布置圖����、生產(chǎn)區(qū)域分布圖。
4.生產(chǎn)過程有凈化要求的���,應(yīng)當(dāng)提供有資質(zhì)的檢測機構(gòu)出具的環(huán)境檢測報告(附平面布局圖)復(fù)印件�。
5.產(chǎn)品生產(chǎn)工藝流程圖��,應(yīng)當(dāng)標(biāo)明主要控制點與項目及主要原材料�����、采購件的來源及質(zhì)量控制方法���。
6.主要生產(chǎn)設(shè)備和檢驗設(shè)備(包括進貨檢驗����、過程檢驗���、出廠最終檢驗所需的相關(guān)設(shè)備����;在凈化條件下生產(chǎn)的,還應(yīng)當(dāng)提供環(huán)境監(jiān)測設(shè)備)目錄�����。
7.質(zhì)量管理體系自查報告��。
8.如適用����,應(yīng)當(dāng)提供擬核查產(chǎn)品與既往已通過核查產(chǎn)品在生產(chǎn)條件、生產(chǎn)工藝等方面的對比說明����。
三��、參考文獻
[1]《醫(yī)療器械監(jiān)督管理條例》(國務(wù)院令第 739 號)[Z].
[2]《醫(yī)療器械注冊與備案管理辦法》(國家市場監(jiān)督管理總局令第 47 號)[Z].
[3]《醫(yī)療器械說明書和標(biāo)簽管理規(guī)定》(國家食品藥品監(jiān)督管理總局令第 6 號)[Z].
[4]《關(guān)于發(fā)布醫(yī)療器械產(chǎn)品技術(shù)要求編寫指導(dǎo)原則的通告》(國家食品藥品監(jiān)督管理總局 2014 年第 9 號通告)[Z].
[5]國家藥監(jiān)局關(guān)于實施《醫(yī)療器械注冊與備案管理辦法》和《體外診斷試劑注冊與備案管理辦法》有關(guān)事項的通知(食藥監(jiān)械管[2021]76 號)[Z].
[6]總局關(guān)于發(fā)布醫(yī)療器械分類目錄的公告(2017 年第104 號)[Z].
[7]國家藥監(jiān)局關(guān)于發(fā)布醫(yī)療器械臨床評價技術(shù)指導(dǎo)原則等 5 項技術(shù)指導(dǎo)原則的通告(2021 年第 73 號)[Z].
[8]國家藥監(jiān)局關(guān)于發(fā)布免于臨床評價醫(yī)療器械目錄的通告(2021 第 71 號)[Z].
[9]《無源手術(shù)器械通用名稱命名指導(dǎo)原則》(國家藥品監(jiān)督管理局通告 2020 年第 79 號)[Z].
[10]GB/T 16886.1����,醫(yī)療器械生物學(xué)評價 第 1 部分:風(fēng)險管理過程中的評價與試驗[S].
[11]GB/T 42062,醫(yī)療器械風(fēng)險管理對醫(yī)療器械的應(yīng)用[S]. [12]YY/T 0466.1����,醫(yī)療器械 用于醫(yī)療器械標(biāo)簽、標(biāo)記和提供信息的符號 第 1 部分:通用要求[S].
四、編寫單位
上海市醫(yī)療器械化妝品審評核查中心