一���、引言
人用基因治療制品通常由含有工程化基因構(gòu)建體的載體或遞送系統(tǒng)組成�����,其活性成分可為核酸(DNA和RNA)�����、基因改造的病毒��、細(xì)菌或細(xì)胞���,通過將外源基因(或基因編輯工具)導(dǎo)入靶細(xì)胞或組織��,替代、補(bǔ)償�����、沉默、修正��、增加或敲除特定基因�����,以達(dá)到治療疾病的目的��。
自20世紀(jì)90年代以來��,基因治療領(lǐng)域的相關(guān)研究呈現(xiàn)持續(xù)上升的趨勢���,多款細(xì)胞治療和基因治療制品獲批上市�����。以重組腺相關(guān)病毒(recombinantAdeno-Associated Virus��,rAAV)載體的基因治療制品為例���,全球已有6款rAAV制品獲批上市,包括Glybera(EMA,2012)��、Luxtura(FDA���,2017)�����、Zolgensma(FDA��,2019)�����、Upstaza(EMA���,2022)、Roctavian(EMA��,2022)和Hemgenix(FDA��,2022)��。目前�����,國內(nèi)也有多達(dá)22款rAAV制品獲得臨床許可,涉及治療眼科疾病�����、血液系統(tǒng)疾病���、神經(jīng)系統(tǒng)疾病及感染性疾病等適應(yīng)證。
基因治療制品中使用的載體通常為重組改構(gòu)載體�����,主要目的是降低臨床安全風(fēng)險(xiǎn)��,提升制品療效�����?�;蛑委熤破酚性S多不同類型��,大體分為3類:① 病毒載體類��,如腺病毒、腺相關(guān)病毒���、慢病毒���、單純皰疹病毒、逆轉(zhuǎn)錄病毒���、痘病毒和仙臺病毒等�����;② 核酸載體類��,如質(zhì)粒載體或基于染色體的載體( 如iBAC��、S/MAR和轉(zhuǎn)座子載體)及RNA等�����;③ 細(xì)菌載體類��,如改良的乳球菌屬�����、減毒李斯特氏菌屬和鏈球菌屬��?�?傇O(shè)計(jì)思路為降低毒性���、增強(qiáng)安全性和提升治療效果�����,具體包括:刪減與毒力�����、致病性或復(fù)制能力相關(guān)的基因、靶向特定組織或細(xì)胞�����、插入細(xì)胞因子基因等增強(qiáng)治療性效果的基因等�����。
另外�����,溶瘤病毒(Oncolytic Virus����,OV)逐漸成為重要發(fā)展方向之一,2015年��,F(xiàn)DA批準(zhǔn)了1款溶瘤病毒制品(Imlygic)上市��。作為一類經(jīng)過基因改造的病毒制品(也可以是野生型)����,在技術(shù)監(jiān)管層面與病毒載體類基因治療制品相似,故合并討論����。
二、概況
基因治療制品種類的多樣性�、特性復(fù)雜性和制造工藝復(fù)雜性都對生產(chǎn)和質(zhì)量控制提出非常高的要求?;蛑委熤破费邪l(fā)品種和開展臨床試驗(yàn)品種逐年增加��。
1����、目的與適用范圍
基因治療制品因起始原材料差異大��、制備工藝不成熟�、生物學(xué)活性以及安全性等質(zhì)量控制難點(diǎn)問題,限制了其自身的快速發(fā)展�。因此,如何提高基因治療制品的安全性��、有效性、質(zhì)量可控性是基因治療制品研發(fā)單位面臨的問題����。
本概述目的是為基因治療制品從業(yè)者科學(xué)��、合理��、合規(guī)地開展工藝開發(fā)����,規(guī)范��、有序地生產(chǎn)和嚴(yán)謹(jǐn)?shù)刭|(zhì)量控制提供指導(dǎo)性建議,主要針對產(chǎn)品申報(bào)上市階段�,為未來規(guī)?�;彤a(chǎn)業(yè)化打下基礎(chǔ)����。產(chǎn)品臨床階段可根據(jù)各階段的研發(fā)特點(diǎn)和研究目的,參考本概述開展與階段相適應(yīng)的研究��。同時(shí),以易懂的方式為從業(yè)者提供必要的質(zhì)量控制基本原則����。
本概述適用范圍主要為基因治療制品����,活性成分包括質(zhì)粒載體��、病毒載體和細(xì)菌載體等�,其中以病毒載體為重點(diǎn),如腺相關(guān)病毒載體、腺病毒載體�、單純皰疹病毒載體等����,也可用于其他適用的病毒類生物制品。
2��、撰寫依據(jù)
本概述針對國內(nèi)基因治療制品行業(yè)的發(fā)展現(xiàn)狀和技術(shù)水平,以尊重基因治療制品特性為前提����,結(jié)合2020年版《中華人民共和國藥典》(以下簡稱《中國藥典》)三部頒布的《人用基因治療制品總論》《體內(nèi)基因治療產(chǎn)品藥學(xué)研究與評價(jià)技術(shù)指導(dǎo)原則(試行)》和《溶瘤病毒產(chǎn)品藥學(xué)研究與評價(jià)技術(shù)指導(dǎo)原則(試行)》等一系列基因治療制品技術(shù)指導(dǎo)原則和法規(guī)要求�,擬對基因治療制品的質(zhì)量控制提出建設(shè)性意見��。參考的其他規(guī)范還包括《藥品生產(chǎn)質(zhì)量管理規(guī)范》(GMP)、《生物制品的生產(chǎn)和檢定用動物細(xì)胞基質(zhì)制備及質(zhì)量控制》《藥用輔料生產(chǎn)質(zhì)量管理規(guī)范》《中國藥典》通則��,以及FDA、EMA和ICH有關(guān)章節(jié)等。
鑒于基因治療制品的復(fù)雜多樣性和相關(guān)質(zhì)量控制研究的不斷深入,本概述廣泛地邀請了國內(nèi)外相關(guān)行業(yè)專家補(bǔ)充完善相關(guān)內(nèi)容��,希望今后能不斷更新和改版����,從而形成更為科學(xué)合理的基因治療制品的質(zhì)量控制策略�,完善藥學(xué)評價(jià)的科學(xué)共識和質(zhì)量控制的規(guī)范��。
3、主要內(nèi)容
基因治療制品的生產(chǎn)和質(zhì)量控制應(yīng)基于質(zhì)量源于設(shè)計(jì)(QbD)的科學(xué)理念��,運(yùn)用風(fēng)險(xiǎn)評估��、全過程控制和全生命周期管理等手段�,以保障基因治療制品安全����、有效的質(zhì)量要求為目的��,并通過對個(gè)生產(chǎn)環(huán)節(jié)的質(zhì)量控制實(shí)現(xiàn)制品生產(chǎn)的穩(wěn)定性和質(zhì)量的一致性��。
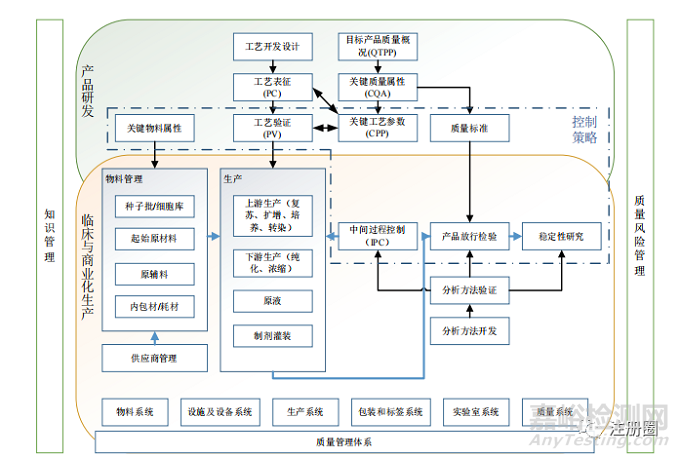
本概述按照基因治療制品的生產(chǎn)工藝順序�,提出生產(chǎn)和質(zhì)量研究需要注意的質(zhì)量控制關(guān)鍵點(diǎn)和風(fēng)險(xiǎn)控制檢查點(diǎn),并提出制品質(zhì)量評價(jià)研究需要開展的內(nèi)容及所用方法�,主要包括:①原輔料選擇、質(zhì)量控制和管理��;②載體種子批制備��、質(zhì)量控制和管理����;③生產(chǎn)用細(xì)胞基質(zhì)制備、質(zhì)量控制和管理;④生產(chǎn)條件保障�、工藝過程控制和工藝驗(yàn)證中的質(zhì)量研究��;⑤制品放行檢驗(yàn)項(xiàng)目質(zhì)量控制研究;⑥制品穩(wěn)定性的質(zhì)量控制等��。詳見圖1��。
▲ 圖1-基因治療制品質(zhì)量控制關(guān)鍵點(diǎn)和風(fēng)險(xiǎn)控制檢查點(diǎn)
4、撰寫原則
基因治療制品質(zhì)量控制需要遵循質(zhì)量源于設(shè)計(jì)(QbD)的理念��,運(yùn)用風(fēng)險(xiǎn)評估的手段����,結(jié)合實(shí)驗(yàn)設(shè)計(jì)(DoE)的工具,明確質(zhì)量控制中關(guān)鍵質(zhì)量屬性(CQA)��,建立有效的質(zhì)量控制體系����,確?���;蛑委熤破返陌踩院陀行?�,保證基因治療制品的生產(chǎn)穩(wěn)定性和質(zhì)量一致性��,盡最大可能降低制品的可變性����,提高基因治療制品質(zhì)量的全生命周期管理水平��。
三����、基因治療制品原輔料及質(zhì)量控制
基因治療制品的生產(chǎn)用物料是指生產(chǎn)過程中所使用的所有生物材料和化學(xué)材料����。按功能分類,主要包括起始原材料�、生產(chǎn)過程中使用或添加的原材料、輔料��、包裝材料和生產(chǎn)用耗材�。起始原材料,一般是指生產(chǎn)中所涉及的生物原材料����,常見的包括質(zhì)粒DNA、細(xì)菌種子批��、病毒種子批�、產(chǎn)毒細(xì)胞株/庫等����,所有相關(guān)起始原材料均應(yīng)進(jìn)行充分的鑒定并建立明確的質(zhì)量控制要求;生產(chǎn)過程中使用或添加的原材料,是指生產(chǎn)中所使用但不作為最終產(chǎn)品成分的物料�,主要包括培養(yǎng)基及其添加成分、工具酶��、緩沖液等��;輔料����,是指生物制品配方中所使用的輔助材料�,如佐劑、穩(wěn)定劑����、賦形劑等;包裝材料�,是指藥品包裝所用的材料,包括與藥品直接接觸的包裝材料和容器�、印刷包裝材料�;生產(chǎn)用耗材��,是指生物制品生產(chǎn)按批次使用的消耗品����,常見的耗材如生物反應(yīng)袋��、儲液袋��、濾器�、膜包等����,也包括層析柱、灌裝組件等�。
基因治療制品的生產(chǎn)用物料與基因治療制品的質(zhì)量����、安全性和有效性密切相關(guān),應(yīng)選擇符合其預(yù)期用途及可穩(wěn)定供應(yīng)的物料����。應(yīng)規(guī)范建立生產(chǎn)用物料的質(zhì)量管理體系����,基于風(fēng)險(xiǎn)進(jìn)行質(zhì)量控制,降低質(zhì)量風(fēng)險(xiǎn)��,保障產(chǎn)品質(zhì)量。企業(yè)可參考《中國藥典》的通則“生物制品生產(chǎn)用原材料及輔料質(zhì)量控制”中的要求來制定物料的管理規(guī)則��。
1、原材料��、輔料��、內(nèi)包材和生產(chǎn)用耗材的質(zhì)量控制
基因治療制品的生產(chǎn)用物料作為制品工藝與質(zhì)量實(shí)現(xiàn)的關(guān)鍵要素之一�,其管理與質(zhì)量控制應(yīng)遵循相關(guān)法規(guī)的要求��,相關(guān)行業(yè)指南也為此提供了指導(dǎo)意見�。2010版GMP第六章“物料與產(chǎn)品”和第十章第七節(jié)“供應(yīng)商的評估和批準(zhǔn)”對物料以及物料供應(yīng)商的管理提出基本要求�。2020年版《中國藥典》三部“生物制品生產(chǎn)用原材料及輔料質(zhì)量控制”通則中對生產(chǎn)過程中使用的原材料和輔料質(zhì)量控制提出了通用性要求����。
生產(chǎn)用物料的質(zhì)量控制活動主要在以下環(huán)節(jié)展開:基于產(chǎn)品關(guān)鍵質(zhì)量屬性以及工藝評估確定物料關(guān)鍵屬性�,篩選確定物料供應(yīng)商��,建立物料與供應(yīng)商的分級分階段管理策略����,建立物料的企業(yè)內(nèi)控質(zhì)量標(biāo)準(zhǔn)與入廠控制策略,實(shí)施入廠后的驗(yàn)收��、檢驗(yàn)、放行����、貯存�、發(fā)放與使用、退庫與銷毀����、物料變更等管理��。
生產(chǎn)過程中使用或添加的原材料����,其質(zhì)量應(yīng)符合其預(yù)期用途�。原材料的選用應(yīng)經(jīng)過充分的工藝開發(fā)��,其來源�、組成��、用途、用量和質(zhì)量等情況應(yīng)明確并合理����,不必要的使用有可能增加殘留的安全性風(fēng)險(xiǎn)和引入外源因子的風(fēng)險(xiǎn)��。關(guān)鍵性原材料優(yōu)先選用藥品監(jiān)管機(jī)構(gòu)批準(zhǔn)的產(chǎn)品或藥用級別的原材料����。應(yīng)充分考慮來源于動物(或人)的原材料可能帶來的外源因子污染的安全性風(fēng)險(xiǎn)�,如有可能,應(yīng)避免選用血清��、豬胰蛋白酶等動物或人來源的原材料����。應(yīng)避免使用毒性較大的化學(xué)原材料��。應(yīng)對選用的原材料的生產(chǎn)地址�、生產(chǎn)工藝、質(zhì)量標(biāo)準(zhǔn)、貨號等建立適當(dāng)?shù)墓芸匾螅瑧?yīng)評估審核其可傳播性海綿體腦炎(TransmissibleSpongiform Encephalopathies and Bovine Spongiform Encephalopathy��,TSE/BSE)風(fēng)險(xiǎn)�、批次出廠檢驗(yàn)報(bào)告等����。應(yīng)基于風(fēng)險(xiǎn)����,對物料進(jìn)行等級評估,評估的因素通常會考慮物料在產(chǎn)品原液、中間產(chǎn)品/半成品和最終制劑等生產(chǎn)制備過程中發(fā)揮的作用����,物料在產(chǎn)品生產(chǎn)過程中的使用或加入步驟,該物料使用或加入后對原液����、制劑或最終產(chǎn)品的影響,以及物料本身的穩(wěn)定性等性質(zhì)��。對于積累了一定物料供應(yīng)歷史情況的企業(yè)�,還應(yīng)考慮物料供應(yīng)商的上市許可、生產(chǎn)����、質(zhì)量控制與供應(yīng)歷史等因素,綜合評估物料的風(fēng)險(xiǎn)等級��。并以此為基礎(chǔ)�,結(jié)合產(chǎn)品研發(fā)或生產(chǎn)所處的不同階段,制定恰當(dāng)?shù)馁|(zhì)量控制策略����,建立合理的企業(yè)內(nèi)控質(zhì)量標(biāo)準(zhǔn)與入廠控制策略,如處于產(chǎn)品臨床前研究階段和非關(guān)鍵臨床階段所使用的原材料和關(guān)鍵臨床階段及商業(yè)化生產(chǎn)階段所使用的物料�,可考慮采用不同的入廠檢驗(yàn)控制策略��。內(nèi)控質(zhì)量標(biāo)準(zhǔn)可參考相關(guān)藥典標(biāo)準(zhǔn)��,并兼顧物料的預(yù)期用途、對產(chǎn)品質(zhì)量的影響��、物料本身的質(zhì)量風(fēng)險(xiǎn)等進(jìn)行評估制訂�。
輔料的選擇、用量和質(zhì)量標(biāo)準(zhǔn)應(yīng)基于制劑的處方開發(fā)研究確定�,證明其使用的必要性、安全性和合理性��。輔料質(zhì)量應(yīng)滿足其預(yù)期功能��。應(yīng)優(yōu)先選用藥品監(jiān)管機(jī)構(gòu)批準(zhǔn)的產(chǎn)品或經(jīng)過注冊備案的�、符合藥用標(biāo)準(zhǔn)的輔料。應(yīng)對選用的輔料的注冊或備案證明����、生產(chǎn)地址、生產(chǎn)工藝����、質(zhì)量標(biāo)準(zhǔn)、貨號等建立適當(dāng)?shù)墓芸匾?�,?yīng)評估審核其TSE/BSE風(fēng)險(xiǎn)、批次出廠檢驗(yàn)報(bào)告等����。由于輔料使用于制劑的生產(chǎn)中,通常被視為關(guān)鍵物料����,其內(nèi)控標(biāo)準(zhǔn)通常參照藥典標(biāo)準(zhǔn),對于早期臨床試驗(yàn)用藥品所用輔料可依據(jù)供應(yīng)商的檢驗(yàn)報(bào)告放行�,但至少應(yīng)當(dāng)通過鑒別或?qū)Φ确绞剑_保其正確無誤��?�;蛑委熕幬镆话銥闊o菌制劑��,其輔料的內(nèi)控標(biāo)準(zhǔn)需要考慮微生物負(fù)載或無菌檢查����、內(nèi)毒素等安全性。
內(nèi)包材的質(zhì)量應(yīng)滿足其預(yù)期功能�。優(yōu)先選用藥品監(jiān)管機(jī)構(gòu)批準(zhǔn)的產(chǎn)品或經(jīng)過注冊備案的、符合藥用標(biāo)準(zhǔn)的內(nèi)包材�。應(yīng)對選用的內(nèi)包材的注冊或備案證明、生產(chǎn)地址����、生產(chǎn)工藝����、質(zhì)量標(biāo)準(zhǔn)�、貨號、批次出廠檢驗(yàn)報(bào)告等建立適當(dāng)?shù)墓芸匾?�。由于?nèi)包材直接與產(chǎn)品接觸�,通常被視為關(guān)鍵物料��。應(yīng)基于風(fēng)險(xiǎn)�,制定恰當(dāng)?shù)馁|(zhì)量控制策略,如處于產(chǎn)品臨床前研究階段和非關(guān)鍵臨床階段所使用的關(guān)鍵包材��,可依據(jù)供應(yīng)商的檢驗(yàn)報(bào)告放行����,但至少應(yīng)當(dāng)通過鑒別或核對等方式,確保其正確無誤����。若采用無菌包裝,還需目測檢查外包裝的完整性以及滅菌標(biāo)識的符合性�;進(jìn)入關(guān)鍵臨床階段后��,對可能引入污染的項(xiàng)目(如可見異物����、微粒��、微生物����、細(xì)菌內(nèi)毒素等)進(jìn)行檢查或控制,還應(yīng)增加一些關(guān)鍵項(xiàng)目的檢驗(yàn)�。關(guān)鍵項(xiàng)目可基于物料的預(yù)期用途、對產(chǎn)品質(zhì)量的影響�、物料本的質(zhì)量風(fēng)險(xiǎn)進(jìn)行評估制訂,具體可參照藥包材國家標(biāo)準(zhǔn)或參考供應(yīng)商提供的標(biāo)準(zhǔn)��。
生產(chǎn)用耗材的使用��,應(yīng)考慮以下幾方面的因素:一是耗材材質(zhì)與工藝的匹配性控制��,如溫度����、耐滅菌性等;二是耗材的動物源性風(fēng)險(xiǎn)����,評估審核其TSE/BSE風(fēng)險(xiǎn)��;三是耗材與產(chǎn)品接觸面(直接接觸的溶液�、生產(chǎn)中間產(chǎn)物等)之間的相互作用����,如化學(xué)相容性、生物相容性����、吸附��、浸出物��、可提取物��、對產(chǎn)品質(zhì)量/穩(wěn)定性的影響�;四是耗材的功能/性能與工藝的匹配性控制,如濾器膜包的過濾效果����、使用條件、一次性系統(tǒng)的組件與功能配置等����。生產(chǎn)用耗材應(yīng)滿足其預(yù)期功能��。應(yīng)對選用的生產(chǎn)用耗材的生產(chǎn)地址�、生產(chǎn)工藝����、質(zhì)量標(biāo)準(zhǔn)、貨號等建立適當(dāng)?shù)墓芸匾?,?yīng)評估審核其TSE/BSE風(fēng)險(xiǎn)�、批次出廠檢驗(yàn)或符合性報(bào)告等�。應(yīng)基于風(fēng)險(xiǎn)��,對耗材進(jìn)行等級評估����,評估的因素通常會考慮耗材在產(chǎn)品原液、中間產(chǎn)品/半成品和最終制劑等生產(chǎn)制備過程中發(fā)揮的作用�,在產(chǎn)品生產(chǎn)過程中的使用步驟,以及該耗材使用后對原液�、制劑或最終產(chǎn)品的影響。同樣����,對于積累了一定物料供應(yīng)數(shù)據(jù)的企業(yè),還應(yīng)考慮供應(yīng)商的上市許可�、生產(chǎn)�、質(zhì)量控制與供應(yīng)歷史等因素,綜合評估耗材的風(fēng)險(xiǎn)等級�。并以此為基礎(chǔ),結(jié)合產(chǎn)品所處的不同階段��,制定恰當(dāng)?shù)馁|(zhì)量控制策略����,建立合理的企業(yè)內(nèi)控質(zhì)量標(biāo)準(zhǔn)與入廠控制策略�。
在早期的基礎(chǔ)研究時(shí),研究者對原材料及輔料的關(guān)注可能不足��,但由于原材料和輔料對制品的質(zhì)量及安全性均有重要影響����,因此��,一旦準(zhǔn)備進(jìn)入制品開發(fā)階段�,建議研發(fā)人員盡早開展原材料及輔料的評估及篩選并制定相應(yīng)的內(nèi)控質(zhì)量標(biāo)準(zhǔn),除了考慮原材料及輔料對工藝的適用性��,還應(yīng)考慮供應(yīng)鏈的風(fēng)險(xiǎn)����,而且在臨床過程中要進(jìn)一步開展相關(guān)的研究,在臨床前應(yīng)完成充分的質(zhì)量評估工作����,建立相對穩(wěn)定的供給和保障體系。
輔助病毒或質(zhì)粒是生產(chǎn)制備一些非復(fù)制型病毒載體的關(guān)鍵材料����,其質(zhì)量及屬性直接影響制品的質(zhì)量和屬性����,應(yīng)根據(jù)應(yīng)用作為起始原材料或者關(guān)鍵原材料來管理�,進(jìn)行更加充分的質(zhì)量研究和質(zhì)量控制。
輔助病毒的質(zhì)量控制要求可參照2020年版《中國藥典》三部“生物制品生產(chǎn)檢定用菌毒種管理及質(zhì)量控制”通則中病毒種子批的相關(guān)要求�����。對用于瞬時(shí)共轉(zhuǎn)染生產(chǎn)過程的質(zhì)粒載體�,需對其來源、特性�、分離純化方法以及核酸序列等進(jìn)行描述,對質(zhì)粒載體的復(fù)制起始點(diǎn)����、啟動子以及編碼選擇性標(biāo)記的基因等組成元件的來源和功能進(jìn)行說明。質(zhì)粒的生產(chǎn)應(yīng)基于細(xì)菌種子批系統(tǒng)���,并符合相關(guān)要求�����。應(yīng)使用適宜的方法純化質(zhì)粒,并基于風(fēng)險(xiǎn)分析和制品特性建立質(zhì)粒載體的質(zhì)量標(biāo)準(zhǔn)����,通常檢測項(xiàng)目包括:鑒別(如限制性酶切圖譜和Sanger測序等)���、質(zhì)粒含量、質(zhì)粒純度��、宿主細(xì)胞DNA殘留量�、質(zhì)粒超螺旋比例或其他可映轉(zhuǎn)染效率的檢測項(xiàng)目、細(xì)菌內(nèi)毒素檢查和無菌檢查或微生物限度檢查(若有必要����,可增加宿主細(xì)胞RNA殘留量、宿主細(xì)胞蛋白質(zhì)殘留量)等���,檢測結(jié)果符合要求后才能用于載體的生產(chǎn)���。
質(zhì)粒載體(或質(zhì)粒DNA)是重要的起始原材料或者關(guān)鍵原材料,建議納入質(zhì)量管理體系進(jìn)行管理���,如建立相應(yīng)的主種子批和工作種子批�����,按照GMP相關(guān)要求生產(chǎn)�,建立質(zhì)量標(biāo)準(zhǔn)并檢驗(yàn),質(zhì)粒的保存和穩(wěn)定性研究可參照生物制品的有關(guān)規(guī)定和指導(dǎo)原則開展��。
3���、原材料及輔料的選擇和風(fēng)險(xiǎn)控制
原材料及輔料的選擇應(yīng)基于風(fēng)險(xiǎn)評估的原則��,通過風(fēng)險(xiǎn)評估從而建立與之相應(yīng)的風(fēng)險(xiǎn)控制策略�����,優(yōu)先選擇安全級別高的原輔料:藥用級優(yōu)先于非藥用級��、優(yōu)先選擇非動物源性物料����,至少需要無TSE/BSE風(fēng)險(xiǎn)申明材料等��。通常會有以下幾種考慮:優(yōu)先選擇已獲得上市許可的生物制品或藥品無菌制劑����;其次,可以選擇已有國家藥品標(biāo)準(zhǔn)�����、取得國家藥品批準(zhǔn)文號并按照中國現(xiàn)行GMP生產(chǎn)的化學(xué)原料藥和藥用級非動物來源的蛋白水解酶等��;再次��,可以選擇按照國家備案管理的非動物源性藥用輔料等����。
若采用自行研制的原材料,例如某種特殊要求的重組蛋白�,不僅需要建立質(zhì)量標(biāo)準(zhǔn),還需要有制備工藝及其工藝驗(yàn)證等數(shù)據(jù)支持��,有的高風(fēng)險(xiǎn)原材料甚至可能還會需要開展動物體內(nèi)的安全性評估���。此外�,這類原材料的檢測要求需要根據(jù)其使用方式���、下游工藝的清除驗(yàn)證數(shù)據(jù)以及潛在風(fēng)險(xiǎn)來確定����。對于研究級別的生物源性的原材料��,不僅要設(shè)置它們的安全性質(zhì)控項(xiàng)目�,如無菌��、內(nèi)毒素�����、支原體��、分枝桿菌(如適用)及外源病毒污染檢查等�����,還要考慮它們的純度���、效價(jià)或?qū)?xì)胞活化、增殖的生物學(xué)效力的質(zhì)控項(xiàng)目����。動物源性材料的質(zhì)量控制,如牛血清����,應(yīng)結(jié)合物料使用需求及風(fēng)險(xiǎn)評估,參考《中國藥典》新生牛血清通則或其他已有的國家標(biāo)準(zhǔn)進(jìn)行原材料質(zhì)量控制及放行��。
根據(jù)對每一種原材料風(fēng)險(xiǎn)評估的結(jié)果建立相應(yīng)的質(zhì)量檢測項(xiàng)目、檢測方法及放行標(biāo)準(zhǔn)��,并在制品研發(fā)過程中不斷分析關(guān)鍵原材料質(zhì)量對制品質(zhì)量的影響�����,并不斷改進(jìn)關(guān)鍵原材料的質(zhì)量要求����。
4�����、AAV載體制備中的關(guān)鍵原材料和輔料
AAV載體制備中的關(guān)鍵原材料及輔料包括培養(yǎng)基����、培養(yǎng)基添加物、血清����、生長因子、細(xì)胞因子���、蛋白酶��、轉(zhuǎn)染試劑�����、核酸酶��、親和純化用配基��、密度梯度介質(zhì)和去垢劑等�,特別強(qiáng)調(diào)質(zhì)粒、病毒種子批和細(xì)胞基質(zhì)也應(yīng)歸屬于關(guān)鍵原材料����。而生物材料的批間差異顯著且難以標(biāo)準(zhǔn)化增加了引入外源性因子的風(fēng)險(xiǎn),進(jìn)而影響生產(chǎn)工藝的重現(xiàn)性或最終產(chǎn)品的質(zhì)量���,因此����,建議盡可能地使用非動物來源材料���,如無血清培養(yǎng)基和重組酶等�����。
鑒于物料對AAV病毒載體生產(chǎn)的重要性����,生產(chǎn)企業(yè)須提供生產(chǎn)過程中使用的所有物料清單以及相應(yīng)物料的質(zhì)量或等級描述,包括物料以及試劑的供應(yīng)商����、來源、質(zhì)量及各物料在工藝中的使用階段(發(fā)酵和純化)等基本信息��。與產(chǎn)品接觸的關(guān)鍵耗材和設(shè)備如發(fā)酵罐�、培養(yǎng)袋��、培養(yǎng)瓶��、色譜基質(zhì)和一次性管道等資料也需要提交�����。同時(shí)���,生產(chǎn)企業(yè)應(yīng)建立確認(rèn)計(jì)劃�,并提供文件證明用于生產(chǎn)的材料符合其預(yù)期用途的標(biāo)準(zhǔn)(包括物料評估報(bào)告�、檢驗(yàn)報(bào)告書等)。
5、基因治療制品中原材料殘留的質(zhì)量控制
對于風(fēng)險(xiǎn)級別較低的原材料�,可評估制備工藝對其去除的能力。對于風(fēng)險(xiǎn)高的原材料除了評估去除能力外�����,若需要�,可在最適工藝階段或終產(chǎn)品中進(jìn)行殘留量檢測的控制并建立控制標(biāo)準(zhǔn),作為工藝相關(guān)雜質(zhì)的一部分進(jìn)行控制�����,如細(xì)胞因子��、生長因子����、蛋白酶(如重組胰蛋白酶)、核酸酶�、轉(zhuǎn)染試劑(如PEI)����、血清及相關(guān)溶劑等。
6���、制劑配方中輔料的質(zhì)量控制
制劑配方中輔料是成品中除主藥外的其他添加物。在基因治療制品中�,輔料一般為人血清白蛋白�、甘露醇�、蔗糖�����、Tween-20、泊洛沙姆等�。根據(jù)ICH相關(guān)規(guī)定�����,制劑配方中輔料若機(jī)理明確��,作用清楚���,不與主藥發(fā)生配伍反應(yīng)及其他副作用風(fēng)險(xiǎn),可不予檢驗(yàn)���;若使用的制劑配方中輔料成分針對該臨床應(yīng)用途徑尚未有明確安全性使用評估結(jié)論或存在其他毒性作用的風(fēng)險(xiǎn),則要將其列入質(zhì)量標(biāo)準(zhǔn)�,規(guī)定其標(biāo)準(zhǔn)范圍���,加以控制。
7��、病毒載體制備過程中牛血清風(fēng)險(xiǎn)的控制
目前,病毒載體制備中仍多采用含血清工藝���,應(yīng)降低牛血清的風(fēng)險(xiǎn)。一方面��,在使用前參照《中國藥典》新生牛血清通則的質(zhì)量要求進(jìn)行牛血清的檢測��,在病毒載體的質(zhì)量控制中進(jìn)行牛血清白蛋白殘留量的檢測�,在最適工藝階段(如未處理收獲液)增加外源病毒因子和特定牛源病毒因子的檢測控制�;另一方面,鼓勵開展病毒載體的低血清以及無血清生產(chǎn)工藝的研究����,如在病毒包裝階段采用低血清培養(yǎng)或無動物源性培養(yǎng)液替代牛血清的工藝研究,也鼓勵基于化學(xué)合成的培養(yǎng)基生產(chǎn)工藝開發(fā)研究��。此外�,在供應(yīng)商審計(jì)方面,應(yīng)建立長效機(jī)制���,監(jiān)控牛血清產(chǎn)區(qū)的疫情等。
四����、病毒載體及質(zhì)粒載體的制備及質(zhì)量控制
載體是基因治療制品的重要組成部分��,是其所攜帶的基因能否發(fā)揮生物學(xué)效應(yīng)的重要依托。病毒載體結(jié)構(gòu)比較復(fù)雜����,至少包括核酸和衣殼蛋白����,有些還包括囊膜等����。如何實(shí)現(xiàn)低風(fēng)險(xiǎn)的穩(wěn)定生產(chǎn)����,是每一個(gè)從業(yè)者關(guān)心的問題。以病毒為載體的基因治療制品的生產(chǎn)工藝種類較多����,并且涉及內(nèi)容復(fù)雜,病毒種子批管理是在基因治療制品起始原材料管理中的重要質(zhì)控點(diǎn),是進(jìn)行低風(fēng)險(xiǎn)�、穩(wěn)定生產(chǎn)的關(guān)鍵措施之一�����。對于質(zhì)粒載體應(yīng)考慮抗生素抗性基因可能給病人帶來的風(fēng)險(xiǎn)和危害,且不得使用氨芐西林抗性基因�����。
1�����、基因治療制品常用的病毒載體
對于以基因修正為主要目的的病毒載體主要是腺相關(guān)病毒(AAV)和慢病毒(LV)。野生型AAV具有免疫原性弱�、幾乎無致病性、感染范圍廣、基因組小、物理性質(zhì)穩(wěn)定等多種特性����,是作為基因治療的理想載體�;慢病毒具有免疫原性低,包裝容量大��、表達(dá)穩(wěn)定等特點(diǎn)�。
對于以殺傷腫瘤為目的的可復(fù)制性病毒載體,目前在臨床研究中已經(jīng)采用的病毒載體類型主要有單純皰疹病毒(HSV)��、腺病毒(AdV)��、痘苗病毒(VV)����、麻疹病毒(MV)、呼腸孤病毒(RV)����、水泡性口炎病毒(VSV)以及新城疫病毒(NDV)等����。
2�、病毒載體在設(shè)計(jì)與構(gòu)建時(shí)的考量
總體而言,病毒載體在設(shè)計(jì)時(shí)應(yīng)基于制品的預(yù)期臨床效果和安全性進(jìn)行考量�。有效性考量應(yīng)基于制品的作用機(jī)制,如目的基因的表達(dá)����,或通過RNA干擾等方式誘導(dǎo)的基因沉默;安全性考量方面��,應(yīng)重點(diǎn)關(guān)注所使用載體的靶向性����、毒力、免疫原性��,以確保其安全性�。
3、病毒種子批建立的必要性
對于復(fù)制型/復(fù)制缺陷型病毒載體�����,為了保證產(chǎn)品生命周期內(nèi)可以持續(xù)生產(chǎn),提升產(chǎn)品的穩(wěn)定性和一致性�,需要建立均一性良好且通過充分表征鑒定的病毒種子批系統(tǒng),并且應(yīng)該采用二級或三級種子批系統(tǒng)(原始種子批���、主種子批和工作種子批)�,使每一支凍存樣品均具有群體代表性和良好的均一性��,而且需要進(jìn)行充分的質(zhì)量檢定�����,從而最大程度地控制污染風(fēng)險(xiǎn)����。因此�,建立種子批系統(tǒng)是病毒載體質(zhì)量一致性的重要保證之一,推薦建立病毒種子批系統(tǒng)����。
4、病毒種子批系統(tǒng)的建立方
病毒種子批系統(tǒng)包括原始毒種種子�����、主種子批以及工作種子批。原始種子相關(guān)信息應(yīng)包括但不限于:毒種詳細(xì)歷史資料���、分離制備過程����、中間體及最終毒種的基因圖譜和注釋�。若毒種構(gòu)建過程中使用過動物原材料應(yīng)保存詳細(xì)清單;若毒種經(jīng)過噬斑法或有限稀釋法純化�����,或來源于DNA或RNA克隆中的恢復(fù)拯救��,均應(yīng)詳細(xì)記錄��。對于建種子批的毒種�����,應(yīng)詳細(xì)記錄制備���、儲存����、維護(hù)以及檢定的相關(guān)信息,主種子批和工作種子批應(yīng)建立穩(wěn)定的制備工藝并進(jìn)行相應(yīng)檢定�����。
5�、種子批的質(zhì)量控制要求
種子批的總體質(zhì)量控制要求為表征與預(yù)期或設(shè)計(jì)一致,應(yīng)具有相應(yīng)功能��、無外源性/內(nèi)源性污染���、在一定代次內(nèi)穩(wěn)定存在等����。同時(shí)�,考慮到不同級別種子批設(shè)立的目的不同��,為了優(yōu)化實(shí)驗(yàn)資源�,使質(zhì)量控制工作更加合理,建議不同級別種子批進(jìn)行不同項(xiàng)目的檢驗(yàn)����。
無論是病毒載體還是輔助病毒,病毒主種子批(MVB)的質(zhì)量控制應(yīng)包括但不限于:鑒別(基因擴(kuò)增��、特異性序列測定和免疫血清學(xué)檢測等)、滴度(感染滴度�、轉(zhuǎn)導(dǎo)滴度等)、活性(治療序列的轉(zhuǎn)錄/表達(dá)量��、治療序列的生物活性)��、無菌檢查(細(xì)菌和真菌)����、支原體檢查、外源病毒因子�、伴生型病毒或復(fù)制型病毒檢查等。其中的病毒外源因子檢查����,除采用傳統(tǒng)藥典方法外,還應(yīng)對原材料�、細(xì)胞基質(zhì)可能引入或高風(fēng)險(xiǎn)的種屬特異性外源病毒因子進(jìn)行篩查。若制品為溶瘤病毒����,還應(yīng)選擇適當(dāng)?shù)姆椒▽Σ《镜哪康幕蜻M(jìn)行序列確證,對殺傷活性以及選擇性增殖活性進(jìn)行測定�。此外,還應(yīng)證明生產(chǎn)用毒種的遺傳穩(wěn)定性、目的基因表達(dá)穩(wěn)定性和生產(chǎn)穩(wěn)定性���。
由于工作種子批(WVB)是由MVB經(jīng)細(xì)胞基質(zhì)擴(kuò)增而來��,在保證細(xì)胞基質(zhì)質(zhì)量可控�����、制備工藝穩(wěn)定的前提下�����,若MVB 進(jìn)行了全面檢定���,WVB 可側(cè)重檢測從 MVB 到 WVB 傳代過程中可能引入的外源因子等;但有特殊需求除外����,如因主種子批數(shù)量不足無法完成外源因子全面檢查時(shí),應(yīng)對工作種子進(jìn)行全面檢定��。
6�����、生產(chǎn)用種子批的管理
2020年版《中國藥典》三部明確規(guī)定了病毒種子批和細(xì)胞庫管理要求�����,包括病毒種子或細(xì)胞一旦從庫中移出不得再回凍至庫內(nèi)�����;研究用種子和細(xì)胞與生產(chǎn)用的種子和細(xì)胞要嚴(yán)格分開存放�;需建立臺賬、凍存容器的監(jiān)測及維護(hù)等���。
7��、病毒種子批的穩(wěn)定性研究
病毒種子的穩(wěn)定性研究數(shù)據(jù)是設(shè)置其保存條件及期間核查周期的重要依據(jù)����,因病毒的生產(chǎn)成本較高�,在研究早期可重點(diǎn)開展保存溫度及不同狀態(tài)的穩(wěn)定性研究。穩(wěn)定性研究主要考查病毒對保存條件敏感的質(zhì)量參數(shù)�,如可見異物、感染滴度等�;若對病毒種子進(jìn)行凍干保存,還需進(jìn)行水分的檢測�����。
種子批的遺傳穩(wěn)定性評價(jià)應(yīng)依據(jù)種子批代次進(jìn)行,因此從主種子開始應(yīng)規(guī)定明確的代次�����。當(dāng)種子批的用途不同時(shí)�,進(jìn)行穩(wěn)定性評估所需要到達(dá)的代次也不同,只用于生產(chǎn)的種子批�����,應(yīng)在生產(chǎn)結(jié)束時(shí)��,仍能保持遺傳穩(wěn)定����;而形成制品用于患者的種子批,應(yīng)考慮在患者體內(nèi)的極限高代次仍應(yīng)具有遺傳穩(wěn)定性����。
種子批的遺傳穩(wěn)定性評價(jià),應(yīng)以遺傳變異的可接受程度為參考依據(jù)�,建立針對多個(gè)高變異檢查點(diǎn)的檢測方法,并且需要建立變異后的可接受標(biāo)準(zhǔn)等�。
8、病毒種子批的外源因子污染檢測
外源因子污染是影響基因治療制品安全性的一個(gè)重要風(fēng)險(xiǎn)因素����,其檢測項(xiàng)目主要包括細(xì)菌、真菌�、支原體及外源病毒因子污染,一般要求不得檢出外源因子污染���。
外源因子污染的檢查方法可參見2020年版《中國藥典》三部通則中無菌檢查法��、支原體檢查法及外源病毒因子污染檢查法���。
在進(jìn)行無菌檢查時(shí),除需進(jìn)行常規(guī)的無菌檢查試驗(yàn)外��,對于培養(yǎng)時(shí)間較長的結(jié)核分桿菌��,需更換專用培養(yǎng)基并延長培養(yǎng)時(shí)間進(jìn)行結(jié)核分枝桿菌的獨(dú)立檢查���。對于支原體檢查���,藥典要求同時(shí)進(jìn)行培養(yǎng)法和DNA染色法檢查,在藥典方法檢測受限時(shí)也可考慮采用特異的熒光定量PCR等替代方法�����。在進(jìn)行病毒類外源因子檢查時(shí),由于制品本身為病毒(尤其是溶瘤病毒制品)�����,為了盡量減少病毒載體自身對試驗(yàn)的干擾��,在開展外源病毒因子檢查(動物試驗(yàn)法或指示細(xì)胞培養(yǎng)法)前����,應(yīng)制備合適的中和抗體或抗血清對制品病毒進(jìn)行中和。若無法制備合適滴度的中和抗體����,需要在病毒種子生產(chǎn)時(shí),設(shè)置生產(chǎn)用對照細(xì)胞并進(jìn)行外源因子檢測����。對于不易培養(yǎng)或風(fēng)險(xiǎn)性較高的外源病毒,如高風(fēng)險(xiǎn)人源病毒�、豬源、猴源��、鼠源病毒等�����,應(yīng)開發(fā)特異性檢測方法,如PCR等分子檢測技術(shù)���。對于逆轉(zhuǎn)錄病毒檢查,一般采用逆轉(zhuǎn)錄酶活性檢查法檢測逆轉(zhuǎn)錄酶活性�����,若逆轉(zhuǎn)錄酶陽性���,還應(yīng)結(jié)合電鏡以及感染性試驗(yàn)結(jié)果來進(jìn)一步判斷是否存在逆轉(zhuǎn)錄病毒���。
除以上方法外,鼓勵研究者開發(fā)靈敏度更高��、簡便快捷的外源因子檢測方法����,如高通量測序(NGS)法、激光力細(xì)胞學(xué)(LFC)法等�,經(jīng)驗(yàn)證后,可考慮作為目前外源病毒因子污染檢查的補(bǔ)充方法�,但此類方法通常檢測的是特定的病毒組分(如核酸序列),可能造成假陽性結(jié)果�,應(yīng)以病毒種子批中是否具有可復(fù)制的外源因子為最終判定依據(jù)。若在測序結(jié)果中測得外源病毒序列��,還需要進(jìn)行PCR驗(yàn)證�,若PCR結(jié)果為陽性,最終仍需要進(jìn)行感染性試驗(yàn)以確證制品中是否存在感染性/可復(fù)制性病毒顆粒���。
9、無中和抗體時(shí)毒種的外源因子檢測
對于桿狀病毒生產(chǎn)系統(tǒng)���,在毒種庫階段進(jìn)行廣譜病毒外源因子���、牛源病毒以及支原體(指示細(xì)胞法)等項(xiàng)目檢查時(shí),需要預(yù)先采用中和抗體對病毒種子批樣品中的主病毒進(jìn)行中和��。當(dāng)中和抗體不能有效中和主病毒時(shí)��,需要進(jìn)行預(yù)稀釋�。為了避免潛在的外源因子因過度稀釋造成的檢驗(yàn)結(jié)果假陰性,預(yù)稀釋倍數(shù)建議不得超過生產(chǎn)接種時(shí)該工藝操作單元毒種的最大放大倍數(shù)?��;诖朔N情況�,一般需要在病毒種子批檢定之前制備高滴度的中和抗體����。
對于無法制備足量高滴度中和抗體的情況����,一方面,可以在毒種制備過程中設(shè)置對照細(xì)胞��,即對照細(xì)胞和毒種培養(yǎng)物同步處理�,但不感染病毒��,最終對對照細(xì)胞進(jìn)行外源因子檢測�����;另一方面�,可以考慮采用經(jīng)過驗(yàn)證的替代方法,如廣譜外源病毒因子檢查可以采用NGS法����,以補(bǔ)充或替代體外細(xì)胞培養(yǎng)以及動物試驗(yàn)用于檢測已知�����、未知或非預(yù)期的病毒種類�。
10�、質(zhì)粒菌種子批的制備要求和質(zhì)控項(xiàng)目
質(zhì)粒是病毒載體生產(chǎn)的起始原材料,而細(xì)菌是質(zhì)粒的宿主����,為了保證質(zhì)粒的穩(wěn)定性和一致性,應(yīng)建立質(zhì)粒菌種子批����。
建立質(zhì)粒菌種子批時(shí)應(yīng)確保菌種子批的歷史和來源清晰,應(yīng)建立至少兩級種子批系統(tǒng)�,應(yīng)確保不存在其他細(xì)菌、真菌和噬菌體的污染��,以及在限定代次內(nèi)具有遺傳穩(wěn)定性����。
菌種子批的檢定通常應(yīng)包括菌落形態(tài)、染色鏡檢、生化特性�、抗生素抗性檢查、電鏡檢查�、質(zhì)粒限制性酶切圖譜分析等。應(yīng)選擇合適的方法證明不存在其他細(xì)菌����、真菌和噬菌體的污染,證明細(xì)菌種子批多批次傳代后基因型和表型的穩(wěn)定性���。對于基因組重要區(qū)域(如人為改造區(qū)域及其側(cè)翼序列)應(yīng)進(jìn)行該區(qū)域的序列測定��,對于經(jīng)基因改造后不大于50 kb的質(zhì)粒,應(yīng)進(jìn)行質(zhì)粒全序列測定�。對于轉(zhuǎn)導(dǎo)質(zhì)粒的細(xì)菌種子批,應(yīng)檢測質(zhì)?����?截悢?shù)和有/無質(zhì)粒細(xì)菌的比例����。對于生產(chǎn)質(zhì)粒用途的細(xì)菌種子批,應(yīng)檢查質(zhì)粒產(chǎn)率�。對于引入的治療基因,應(yīng)檢測其表達(dá)和功能活性。對于減毒細(xì)菌載體����,應(yīng)鑒定其減毒的特性和穩(wěn)定性,并檢測其對抗生素的敏感性�。
11、生產(chǎn)用質(zhì)粒載體的質(zhì)量控制項(xiàng)目
根據(jù)2020年版《中國藥典》三部“人用基因治療制品總論”中的相關(guān)要求�����,生產(chǎn)用質(zhì)粒DNA載體應(yīng)考慮抗生素抗性基因的風(fēng)險(xiǎn)��,且不得使用氨芐西林抗性基因���。對生產(chǎn)用質(zhì)粒載體的質(zhì)量控制項(xiàng)目���,主要包括鑒別,即對質(zhì)粒改造部位的PCR檢測或?qū)|(zhì)粒特定限制性酶切圖譜的檢測�、對全質(zhì)粒的Sanger測序,隨著測序技術(shù)的發(fā)展����,建議質(zhì)粒全長測序;純度�,通常采用 A260/A280����;含量����,即質(zhì)粒的濃度(A260);質(zhì)粒形態(tài)����,即超螺旋比例(如瓊脂糖凝膠電泳,毛細(xì)管凝膠電泳或AEX-HPLC等)��;建議對質(zhì)粒進(jìn)行微生物限度檢查或無菌檢查���,有條件的推薦無菌檢查���,以保證生產(chǎn)過程較低的微生物負(fù)荷或達(dá)到無菌要求��,若有必要可增加雜質(zhì)檢測項(xiàng)目���,如宿主細(xì)胞DNA�����、RNA以及蛋白質(zhì)殘留量���。
以重組AAV病毒三質(zhì)粒生產(chǎn)系統(tǒng)為例���,用于生產(chǎn)的3個(gè)質(zhì)粒分別為載體質(zhì)粒(AAV-GOI,插入目的基因����,包裝進(jìn)入重組AAV病毒),包裝質(zhì)粒(AAV-RC��,含有AAV復(fù)制相關(guān)的rep基因和衣殼cap基因)和輔助質(zhì)粒(pHelper�,包含腺病毒的E2A、E4和VARNA基因�����,與293細(xì)胞中的E1a和E1b基因共同替代5型腺病毒的作用����,輔助重組AAV病毒的包裝)。對3種質(zhì)粒的質(zhì)量控制項(xiàng)目應(yīng)包括鑒別����、純度��、含量和質(zhì)粒形態(tài)��。對于鑒別����,因?yàn)?種質(zhì)粒所攜帶基因不同����,采用PCR進(jìn)行鑒別時(shí)�,目的片段種類及大小應(yīng)各異����,限制性酶切圖譜鑒別中所用的限制性內(nèi)切酶和酶切圖譜也應(yīng)分別考慮,測序鑒別的結(jié)果應(yīng)符合各質(zhì)粒的預(yù)期序列���。值得注意的是�����,載體質(zhì)粒中ITR區(qū)容易丟失,這會嚴(yán)重影響重組AAV的包裝效率�����,由于ITR區(qū)是反向互補(bǔ)序列,存在高級結(jié)構(gòu)����,PCR不易擴(kuò)增,可采用SmaI限制性酶切后�����,以電泳的方式檢測ITR區(qū)的丟失情況�����。另外�,超螺旋結(jié)構(gòu)的質(zhì)粒形態(tài)具有最好的包裝效率,3種質(zhì)粒均應(yīng)保證較高的超螺旋占比�����,具體可采用瓊脂糖凝膠電泳�����、毛細(xì)管凝膠電泳或高效液相色譜(HPLC)進(jìn)行控制����。
五、細(xì)胞基質(zhì)的制備及質(zhì)量控制
細(xì)胞基質(zhì)是病毒類基因治療制品生產(chǎn)的重要起始原材料之一�,是核酸復(fù)制、衣殼蛋白表達(dá)�����、病毒顆粒組裝的重要場所���。為了保證目標(biāo)制品質(zhì)量的一致性和穩(wěn)定性�,在滿足建庫需求的情況下�����,生產(chǎn)/包裝用細(xì)胞基質(zhì)均應(yīng)建庫管理�。細(xì)胞庫無污染、可穩(wěn)定生產(chǎn)�����,是保障終制品質(zhì)量的基本前提��。在生物污染方面�����,首先要從源頭上控制細(xì)胞種屬來源的外源因子�,其次要保障細(xì)胞庫制備過程中不受外源因子污染。在穩(wěn)定性考察方面���,要重點(diǎn)關(guān)注生產(chǎn)產(chǎn)品的一致性和貯存在規(guī)定條件下的細(xì)胞能否維持其生產(chǎn)能力�。
1�、病毒類基因治療產(chǎn)品的生產(chǎn)用細(xì)胞基質(zhì)
細(xì)胞基質(zhì)是生產(chǎn)病毒類基因治療產(chǎn)品必不可少的起始原材料,一般包括病毒種子批構(gòu)建時(shí)使用的細(xì)胞基質(zhì)和生產(chǎn)病毒過程中使用的細(xì)胞基質(zhì)��,2種細(xì)胞基質(zhì)可能相同也可能不同�����。以昆蟲細(xì)胞-桿狀病毒系統(tǒng)生產(chǎn)rAAV為例����,包括制備桿狀病毒種子批的細(xì)胞基質(zhì)和生產(chǎn)rAAV的細(xì)胞基質(zhì)。
2��、生產(chǎn)用細(xì)胞基質(zhì)的選擇
細(xì)胞基質(zhì)的選擇和使用應(yīng)具有一定的依據(jù)并符合相應(yīng)的生產(chǎn)要求����,任何細(xì)胞基質(zhì)均應(yīng)滿足來源清晰、歷史培養(yǎng)過程清楚、風(fēng)險(xiǎn)可控及經(jīng)過全面檢定的要求���,以確保其適用性和安全性��。在選擇細(xì)胞基質(zhì)前應(yīng)進(jìn)行研究和評估�����,一般包括細(xì)胞的種屬及組織來源����、細(xì)胞對病毒的敏感性及穩(wěn)定生產(chǎn)病毒的能力��、細(xì)胞的特性及全面檢定的可行性����、細(xì)胞對制品的安全性�、生產(chǎn)工藝的便利性和可行性以及下游純化工藝能夠去除風(fēng)險(xiǎn)因素的可能性等。除考慮細(xì)胞生物屬性(如生長特性��、包裝效率等)對病毒產(chǎn)量的影響外�,應(yīng)全面評估細(xì)胞對最終制品質(zhì)量和安全性的潛在影響����,如細(xì)胞是否含有致癌基因��、細(xì)胞成瘤性和致瘤性、內(nèi)源性病毒的污染,以及病毒載體在細(xì)胞內(nèi)的重組風(fēng)險(xiǎn)等。
3���、細(xì)胞庫的建立和管理
為了保證目標(biāo)制品質(zhì)量的一致性和穩(wěn)定性,在滿足建庫需求的情況下,生產(chǎn)/包裝用細(xì)胞基質(zhì)均應(yīng)建庫管理。通常細(xì)胞庫包括主細(xì)胞庫和工作細(xì)胞庫���,參照2020年版《中國藥典》三部“生物制品生產(chǎn)檢定用動物細(xì)胞基質(zhì)制備及質(zhì)量控制”和 ICHQ5D的相關(guān)要求進(jìn)行建庫、保存和管理���。
4��、細(xì)胞培養(yǎng)過程中的注意事項(xiàng)
細(xì)胞取材�����、建庫及制備全過程應(yīng)具有可溯源性及操作的一致性,并對各個(gè)環(huán)節(jié)的風(fēng)險(xiǎn)進(jìn)行充分的評估�。所有類型細(xì)胞的供體應(yīng)無傳染性疾病或未知病原體的疾病����。與細(xì)胞培養(yǎng)相關(guān)的所有材料,特別是人源或動物源性材料��,均應(yīng)按照藥典相關(guān)要求進(jìn)行風(fēng)險(xiǎn)評估��,選擇與生產(chǎn)相適應(yīng)的原材料���,必要時(shí)進(jìn)行檢測��。所有生物源性材料均應(yīng)無細(xì)菌�、真菌�、分枝桿菌����、支原體及病毒等外源因子污染����。細(xì)胞培養(yǎng)過程中所用的牛血清及胰酶應(yīng)符合藥典相關(guān)要求����。細(xì)胞培養(yǎng)液中不得含有人血清�。若使用人血白蛋白���,應(yīng)獲得國家藥品監(jiān)督管理部門批準(zhǔn)�。細(xì)胞制備過程中不得使用青霉素或β-內(nèi)酰胺類抗生素。配制各種溶液的化學(xué)藥品應(yīng)符合《中國藥典》或其他相關(guān)國家標(biāo)準(zhǔn)的要求�。
生產(chǎn)過程中,從凍存的 WCB 中取出1支或多支混合后培養(yǎng)��,傳至一定代次后供生產(chǎn)用�����,其代次不得超過該細(xì)胞用于生產(chǎn)的最高限定代次。生產(chǎn)用細(xì)胞的最高限定代次應(yīng)根據(jù)研究結(jié)果確定����,但不得超過國際認(rèn)可的最高限定代次。從WCB取出的細(xì)胞經(jīng)增殖后獲得的細(xì)胞不得再回凍保存用于生產(chǎn)��。
病毒生產(chǎn)過程中應(yīng)重點(diǎn)關(guān)注外源因子的污染和交叉污染����、生產(chǎn)人員的健康和安全防護(hù)����、生產(chǎn)環(huán)境的生物等級和安全隔離,以及生產(chǎn)產(chǎn)物或廢棄對環(huán)境的生物安全性影響��。
5����、生產(chǎn)用細(xì)胞基質(zhì)的檢驗(yàn)依據(jù)及內(nèi)容
生產(chǎn)用細(xì)胞基質(zhì)的檢定應(yīng)符合2020年版《中國藥典》三部“人用基因治療制品總論”�、通則“生物制品生產(chǎn)檢定用動物細(xì)胞基質(zhì)制備及質(zhì)量控制”�、ICH Q5D的相關(guān)要求,以及國家藥品監(jiān)督管理局藥品審評中心(Center for Drug Evaluation�,CDE)發(fā)布的相關(guān)指導(dǎo)原則。檢定項(xiàng)目包括:鑒別�、微生物安全、內(nèi)外源病毒因子檢測�、成瘤性/致瘤性��、插入序列的鑒別和完整性確認(rèn)(如適用)等���,需關(guān)注細(xì)胞種屬相關(guān)病毒和培養(yǎng)過程可能引入的潛在外源因子的污染風(fēng)險(xiǎn)���。必要時(shí)還須進(jìn)行細(xì)胞生長特性�����、細(xì)胞染色體檢查����,細(xì)胞均一性及穩(wěn)定性檢查。一般根據(jù)細(xì)胞類型、制品的性質(zhì)以及生產(chǎn)工藝�����,決定是否有必要進(jìn)行染色體檢查和成瘤性和/或致瘤性試驗(yàn)���。經(jīng)基因修飾建立的穩(wěn)定傳代細(xì)胞系�,還應(yīng)對基因修飾的結(jié)果��,如基因序列��、修飾位點(diǎn)��、拷貝數(shù)����、表達(dá)水平等進(jìn)行研究確認(rèn)。
6�、MCB、WCB����、EOPC、對照細(xì)胞的檢驗(yàn)
細(xì)胞庫建立后應(yīng)至少對 MCB 細(xì)胞及生產(chǎn)終末細(xì)胞 (EOPC) 進(jìn)行一次全面檢定���,對于生產(chǎn)終末細(xì)胞不易取到的情況��,可采用未加工細(xì)胞收獲液結(jié)合限定代次細(xì)胞的做法進(jìn)行全面檢定��。當(dāng)生產(chǎn)工藝發(fā)生改變時(shí)��,應(yīng)重新對 EOPC 進(jìn)行檢測��。每次從MCB建立一個(gè)新的WCB, 均應(yīng)按規(guī)定項(xiàng)目進(jìn)行檢定����。當(dāng)MCB進(jìn)行全面檢定時(shí),WCB需檢測的外源病毒種類可主要考慮從MCB到WCB傳代過程中可能引入的外源因子�����,而僅存在于MCB建庫前的外源因子可不再重復(fù)檢測����。若在MCB或WCB中未檢測到種屬特異性病毒,后續(xù)過程中可不再進(jìn)行重復(fù)檢測�����。
對照細(xì)胞一般是在病毒種子或制品的外源病毒因子檢查受限的情況下設(shè)置的�。對照細(xì)胞培養(yǎng)物應(yīng)與生產(chǎn)培養(yǎng)物同時(shí)處理,但不感染病毒���。對照細(xì)胞培養(yǎng)物需要在生產(chǎn)條件下再培養(yǎng)14天以上��;培養(yǎng)期間可以換液��,以檢測潛伏感染的��、內(nèi)源的或復(fù)制能力差的潛在病毒因子�����。在不同生產(chǎn)階段�,檢測對照細(xì)胞的外源病毒因子�����。一般在生產(chǎn)末期��,取不接種病毒的對照細(xì)胞�,按藥典相關(guān)規(guī)定進(jìn)行細(xì)胞鑒別試驗(yàn),細(xì)菌�����、真菌無菌檢查,支原體檢查�����,以及病毒外源因子檢查����。
7、細(xì)胞基質(zhì)內(nèi)外源因子檢測的項(xiàng)目和方法
外源因子(Adventitious Agents)指生產(chǎn)中偶然引入的微生物����,一般包括細(xì)菌、真菌���、支原體/螺原體��、分枝桿菌�、立克次體����、病毒、原生動物����、寄生蟲、傳染性海綿狀腦病相關(guān)因子等���。根據(jù)污染途徑分為源頭污染(細(xì)胞種子來源����,如種屬相關(guān)�����、供體相關(guān))和過程污染(培養(yǎng)過程引入��,如污染的培養(yǎng)試劑�����、耗材�、容器、環(huán)境或不當(dāng)操作)��。對于細(xì)胞種屬來源相關(guān)的外源因子����,如人源的HEK293細(xì)胞需要檢測人源病毒因子、猴源的Vero細(xì)胞需要檢測猴源的病毒因子��、昆蟲細(xì)胞系需要考慮昆蟲來源的病毒因子、含有內(nèi)源性病毒的細(xì)胞需進(jìn)行相應(yīng)的控制���;對于某些含有內(nèi)源性逆轉(zhuǎn)錄酶或逆轉(zhuǎn)錄病毒序列的細(xì)胞���,需要采用感染性試驗(yàn)判斷是否有感染性病毒顆粒存在。對于生產(chǎn)過程中可能引入的外源因子���,如采用了動物源性原材料牛血清�、豬胰蛋白酶等�,則需要檢測相對應(yīng)的動物源性病毒因子。
檢測方法可分為體內(nèi)法和體外法����。采用體內(nèi)法時(shí),應(yīng)考慮不同的試驗(yàn)動物對外源因子敏感性的不同���,如成年小鼠對淋巴細(xì)胞性脈絡(luò)叢腦膜炎病毒(LCMV)�、柯薩奇病毒����、蟲媒病毒、狂犬病毒敏感;乳鼠對多種人源病毒敏感�,如柯薩奇病毒A/B和其他小RNA病毒、甲病毒屬�、布尼亞病毒、沙粒病毒�、黃病毒屬�、狂犬病毒和皰疹病毒;豚鼠對結(jié)核桿菌和包括副粘病毒屬����、呼腸孤病毒和絲狀病毒屬在內(nèi)的外源病毒因子敏感;家兔對乙型皰疹病毒敏感����;雞胚尿囊液對正粘病毒、副粘病毒���、甲病毒屬�、水皰病毒屬敏感�����,雞胚卵黃囊對皰疹病毒�、痘病毒、彈狀病毒、立克次體����、支原體和細(xì)菌敏感。若存在可能暴露于某一種屬病毒的風(fēng)險(xiǎn)時(shí)��,應(yīng)根據(jù)種屬來源選用合適的模型進(jìn)行抗體生產(chǎn)試驗(yàn)���。
體外法包括非特異性和特異性的檢測方法����。非特異性方法包括細(xì)胞培養(yǎng)法�、掃描電鏡法,以及逆轉(zhuǎn)錄病毒的非特異檢測方法�。細(xì)胞培養(yǎng)法可以檢測多種非特異性病毒因子,包括致細(xì)胞病變病毒��、血吸附病毒和血凝病毒。指示細(xì)胞的選擇可參考藥典相關(guān)規(guī)定�,一般包括與生產(chǎn)細(xì)胞同種屬同組織來源的細(xì)胞、人二倍體細(xì)胞和猴源細(xì)胞����,對于生產(chǎn)用昆蟲細(xì)胞�,還應(yīng)包括對蟲媒病毒普遍易感的細(xì)胞(如BHK21)�����。掃描電鏡法可以直接觀察病毒顆粒�,盡管靈敏度不高,但是可以檢測多種類型外源因子�。對于逆轉(zhuǎn)錄病毒的非特異性檢測方法可參考藥典相關(guān)規(guī)定。特異性檢測方法主要是基于高靈敏度的方法(如PCR)����,對非特異性檢測方法不易檢出�����、高風(fēng)險(xiǎn)的病毒采用特異性檢測方法,可提高檢測靈敏度���,最大程度地降低生物安全風(fēng)險(xiǎn)�。應(yīng)根據(jù)細(xì)胞種屬來源、組織來源及供體健康狀況等確定檢測病毒的種類����。如人源的細(xì)胞應(yīng)考慮檢測如人EB病毒��、人巨細(xì)胞病毒(HCMV)�����、人逆轉(zhuǎn)錄病毒(HIV-1/2、HTLV-1/2)��、人肝炎病毒(HAV��、HBV�����、HCV)�����、人細(xì)小病毒B19��、人乳頭瘤病毒、人多瘤病毒�,以及難培養(yǎng)的人腺病毒和人皰疹病毒-6/7/8 等���。也可采用非特異性和特異性方法相結(jié)合的方式�,如采用細(xì)胞培養(yǎng)結(jié)合熒光抗體染色法檢測非特異性和特異性牛源病毒����。
NGS技術(shù)已被證明有能力進(jìn)行廣譜的外源病毒檢測�,可用于檢測存在于細(xì)胞DNA中的病毒序列或在細(xì)胞中以RNA表達(dá)的病毒序列���,但應(yīng)進(jìn)行充分的方法學(xué)驗(yàn)證�,包括使用合適的標(biāo)準(zhǔn)品或參照品進(jìn)行分析鑒定和驗(yàn)證,以評估方法性能�,并證明病毒檢測的靈敏度、特異性和廣度。對NGS獲得的陽性結(jié)果應(yīng)進(jìn)一步確定是否與感染性病毒相關(guān)�。
8��、細(xì)胞基質(zhì)的成瘤性和致瘤性評價(jià)
成瘤性是指待檢細(xì)胞接種動物后��,在動物體內(nèi)形成(腫)瘤的特性��。成瘤性檢查的目的是評估待檢細(xì)胞在受試人體內(nèi)形成腫瘤的風(fēng)險(xiǎn)。新建細(xì)胞系/株及新型細(xì)胞基質(zhì)應(yīng)進(jìn)行成瘤性檢查���。某些傳代細(xì)胞系已證明在一定代次內(nèi)不具有成瘤性�����,而超過一定代次則具有成瘤性����,如 Vero 細(xì)胞��,因此�,必須進(jìn)行成瘤性檢查。已證明具有成瘤性的傳代細(xì)胞,如BHK21���、CHO��、HEK293等用于生產(chǎn)治療性制品時(shí)�����,可不再做成瘤性檢查�。
致瘤性是指將待檢細(xì)胞的細(xì)胞成分接種動物后���,誘導(dǎo)動物本身細(xì)胞形成腫瘤的特性�����。致瘤性檢查的目的是評估待檢細(xì)胞DNA和碎片誘導(dǎo)受試人細(xì)胞形成原發(fā)腫瘤的風(fēng)險(xiǎn)����。已建株的二倍體細(xì)胞���,如MRC-5��、2BS��、KMB17�、WI-38及FRhL-2新建主細(xì)胞庫不要求進(jìn)行致瘤性檢查��。已建株的或有充分應(yīng)用經(jīng)驗(yàn)的連續(xù)傳代細(xì)胞����,如CHO、NSO�、Sp2/0、低代次的Vero細(xì)胞不要求進(jìn)行致瘤性檢查����。缺少充分應(yīng)用經(jīng)驗(yàn)的細(xì)胞基質(zhì)需進(jìn)行致瘤性檢查。若使用具有成瘤性的細(xì)胞���,需結(jié)合臨床風(fēng)險(xiǎn)獲益�、給藥途徑和生產(chǎn)工藝的雜質(zhì)去除性能(如活細(xì)胞殘留�、致瘤基因片段的殘留等)等評估其使用的必要性、合理性和安全性����,分析細(xì)胞中是否攜帶具有致瘤風(fēng)險(xiǎn)的基因或其他因子,必要時(shí)���,應(yīng)對其殘留水平和基因片段大小進(jìn)行控制���。一般不建議使用具有致瘤性的細(xì)胞��。另外��,需通過生產(chǎn)過程控制和/或產(chǎn)品放行對完整細(xì)胞��,尤其是成瘤性細(xì)胞的殘留水平進(jìn)行控制���。
9、基因修飾細(xì)胞基質(zhì)的特殊要求
對于經(jīng)基因修飾(如組成性表達(dá)病毒包裝蛋白或復(fù)制輔助因子)的生產(chǎn)用細(xì)胞�����,需開展基因修飾的相關(guān)研究����,包括基因修飾時(shí)所采用質(zhì)粒、病毒����、基因元件的質(zhì)量控制研究資料。細(xì)胞庫的表征研究中還應(yīng)對基因修飾的結(jié)果��,如基因序列、修飾位點(diǎn)���、拷貝數(shù)����、表達(dá)水平等進(jìn)行確認(rèn)��,以確保制品的安全性和有效性��。同時(shí)���,還要評價(jià)細(xì)胞在限定代次內(nèi)的遺傳穩(wěn)定性和功能穩(wěn)定性,如修飾序列的拷貝數(shù)����、表達(dá)產(chǎn)物量、包裝病毒的產(chǎn)量及質(zhì)量等�。應(yīng)考慮基因修飾的必要性和修飾方法的適用性,修飾過程不應(yīng)增加外源因子引入的風(fēng)險(xiǎn)�,修飾基因的選擇應(yīng)盡量避免或降低病毒包裝過程發(fā)生重組的風(fēng)險(xiǎn)。若修飾過程中引入了病毒序列或轉(zhuǎn)化序列��,應(yīng)對潛在風(fēng)險(xiǎn)進(jìn)行充分評估����,如是否存在重組病毒的風(fēng)險(xiǎn)��、細(xì)胞成瘤性和致瘤性的變化�,并在終制品中對序列殘留進(jìn)行控制�����。
10�����、存在安全性風(fēng)險(xiǎn)細(xì)胞的質(zhì)量控制
對于腫瘤細(xì)胞或攜帶致瘤表型�、病毒序列的細(xì)胞,應(yīng)謹(jǐn)慎選用��,若確因病毒生產(chǎn)需要使用��,應(yīng)采用更嚴(yán)格的宿主細(xì)胞核酸殘留限度��,并對完整細(xì)胞的殘留進(jìn)行控制���。在制品放行檢驗(yàn)中��,除了控制宿主細(xì)胞DNA殘留量和大小����,還應(yīng)對已知具有安全性風(fēng)險(xiǎn)的特定轉(zhuǎn)化序列殘留進(jìn)行控制,如293T細(xì)胞的E1A�����、SV40大T抗原��、Hela細(xì)胞的E6/E7基因���,檢測方法應(yīng)具備足夠的靈敏度和特異性。
對于含有內(nèi)源性病毒的細(xì)胞���,原則上不用于生產(chǎn)�,若確因病毒生產(chǎn)需要使用��,應(yīng)評估其使用的必要性和安全性�,如內(nèi)源病毒的人體感染活性、免疫原性��、工藝殘留水平等��,必要時(shí)����,應(yīng)在工藝中增加經(jīng)驗(yàn)證的病毒去除/滅活工藝單元��,并在適當(dāng)?shù)碾A段對內(nèi)源病毒的殘留和活性進(jìn)行檢測�����,以確保不會對成品的安全性�、有效性和質(zhì)量可控性造成影響���。
對于非哺乳動物來源的生產(chǎn)用細(xì)胞����,除了種屬相關(guān)的外源因子�,還要考慮到宿主的種屬差異可能會對病毒衣殼蛋白的表達(dá)比例及翻譯后修飾情況產(chǎn)生影響,建議加強(qiáng)終產(chǎn)品的結(jié)構(gòu)確證�、免疫原性和體內(nèi)生物分布的研究。
11��、細(xì)胞基質(zhì)的穩(wěn)定性評價(jià)
傳代穩(wěn)定性研究的目的是為了確保細(xì)胞基質(zhì)可以穩(wěn)定地生產(chǎn)預(yù)期質(zhì)量一致的制品和貯存在規(guī)定條件下的細(xì)胞可維持其生產(chǎn)能力��。
細(xì)胞庫的傳代穩(wěn)定性研究條件應(yīng)代表或模擬商業(yè)化生產(chǎn)工藝����,重點(diǎn)考察細(xì)胞基質(zhì)的遺傳穩(wěn)定性�����、生產(chǎn)穩(wěn)定性(如染色體檢查�、細(xì)胞生長特性等)�、安全性(外源因子污染和成瘤性、致瘤性變化)�,一般應(yīng)包括鑒別、微生物安全(如無菌���、支原體等)�、內(nèi)/外源病毒因子��、特定外源病毒因子(若適用)����、逆轉(zhuǎn)錄病毒��、染色體檢查(二倍體細(xì)胞)����、細(xì)胞生長特性、生產(chǎn)/包裝病毒能力和病毒產(chǎn)品質(zhì)量(如產(chǎn)物的完整性和一致性)等�。同時(shí)�,需考慮細(xì)胞庫隨著代次增加引起的成瘤性和/或致瘤性變化�。經(jīng)基因修飾建立的細(xì)胞系/株的傳代穩(wěn)定性研究還需關(guān)注傳代過程中基因修飾部分的穩(wěn)定性,如基因序列�����、拷貝數(shù)�����、蛋白表達(dá)穩(wěn)定性��。根據(jù)傳代穩(wěn)定性研究結(jié)果制定細(xì)胞庫限傳代次�����,用于生物制品生產(chǎn)的細(xì)胞最高限定代次須經(jīng)批準(zhǔn)�,實(shí)際生產(chǎn)過程中應(yīng)在細(xì)胞限傳代次內(nèi)進(jìn)行病毒產(chǎn)品生產(chǎn)。
為了保證細(xì)胞庫在貯存過程中的穩(wěn)定性�,需制定合理的貯存穩(wěn)定性考察方案,關(guān)注在長期貯存過程中細(xì)胞活率��、細(xì)胞生長特性�����、生產(chǎn)病毒能力等的變化。根據(jù)研究結(jié)果確定細(xì)胞庫的貯存條件�,細(xì)胞庫在擬定的貯存條件下可滿足生產(chǎn)需求。
六����、基因治療制品生產(chǎn)過程中的質(zhì)量控制
本章節(jié)主要針對生產(chǎn)過程中的質(zhì)量控制,包括生產(chǎn)工藝研究����、中間過程控制、檢查項(xiàng)目���、質(zhì)量標(biāo)準(zhǔn)和工藝驗(yàn)證等���。病毒載體類基因治療制品的生產(chǎn)工藝種類較多,并且涉及內(nèi)容復(fù)雜����。對制品特性的全面而準(zhǔn)確的分析研究����,對已有的、成熟的、有效的生產(chǎn)管理模式的借鑒以及現(xiàn)有的技術(shù)和質(zhì)量管理理念的落實(shí)等�,都是制品生產(chǎn)工藝設(shè)計(jì)和質(zhì)量保障的重要基石。
病毒載體類基因治療制品生產(chǎn)中的質(zhì)量源于設(shè)計(jì)(QbD)和全過程質(zhì)量控制是非常重要的理念��,任何環(huán)節(jié)的疏漏����,都有可能導(dǎo)致災(zāi)難性后果,因此�����,樹立“以制品質(zhì)量為中心的生產(chǎn)意識”尤為重要�。生產(chǎn)過程中各種生產(chǎn)參數(shù)控制是質(zhì)量控制的主體,全過程關(guān)鍵工藝參數(shù)控制和中間過程控制檢查共同組成制品的生產(chǎn)過程質(zhì)量控制�����??刂撇呗钥砂?個(gè)方面:1)中間過程控制樣品檢測;2)工藝參數(shù)的控制�;3)工藝性能的監(jiān)控;4)產(chǎn)品表征�;5)產(chǎn)品放行檢測;6)產(chǎn)品穩(wěn)定性考察�;7)物料控制����;8)廠房設(shè)施和設(shè)備控制����。
1、基因治療制品生產(chǎn)工藝設(shè)計(jì)/研究的考慮因素
生產(chǎn)工藝設(shè)計(jì)和研究是質(zhì)量體系的重要組成部分��。由于基因治療制品的復(fù)雜性和特殊性��,要求基于制品的目標(biāo)產(chǎn)品質(zhì)量特性�,開展生產(chǎn)工藝設(shè)計(jì)/研究,并運(yùn)用風(fēng)險(xiǎn)評估工具完善工藝開發(fā)����,工藝設(shè)計(jì)/研究應(yīng)貫穿基因治療制品的全生命周期中,并隨著技術(shù)的進(jìn)步和認(rèn)識的深入����,不斷補(bǔ)充和完善。
病毒載體類基因治療制品的生產(chǎn)工藝設(shè)計(jì)應(yīng)基于制品特性��,除考慮安全性外����,還要關(guān)注起始生物原材料的遺傳穩(wěn)定性,如適當(dāng)?shù)木痉N種子批和細(xì)胞庫管理模式��,可以有效地控制生產(chǎn)中起始生物原材料遺傳變異所帶來的制品質(zhì)量風(fēng)險(xiǎn)�。
生產(chǎn)過程中原則上應(yīng)盡力避免使用有毒有害的物質(zhì),若不得不使用有毒有害物質(zhì)�,應(yīng)對這些物質(zhì)的殘留進(jìn)行控制。
除此之外���,在制品生產(chǎn)工藝設(shè)計(jì)/研究中還必須滿足法規(guī)的強(qiáng)制要求�,如《中國藥典》凡例中關(guān)于抗生素的使用要求��。
2����、病毒載體類基因治療制品生產(chǎn)過程的關(guān)鍵工藝研究
總體上講,病毒載體基因治療制品的生產(chǎn)步驟主要包括:細(xì)胞基質(zhì)培養(yǎng)�、接種毒種或轉(zhuǎn)染質(zhì)粒、病毒收獲澄清/過濾���、純化和濃縮工藝����、原液存儲和檢測���、成品配制灌裝等��,不同的制品特性工藝存在一定差異���。
細(xì)胞基質(zhì)應(yīng)符合細(xì)胞庫的管理要求����,在向生產(chǎn)容器中接種細(xì)胞基質(zhì)前����,對容器的清潔、培養(yǎng)基的除菌�����、細(xì)胞的復(fù)蘇和擴(kuò)增都非常重要�;在向生產(chǎn)容器中接種細(xì)胞基質(zhì)的過程中,應(yīng)做好防污染的防護(hù)措施�;細(xì)胞基質(zhì)的培養(yǎng)需要對相關(guān)設(shè)備進(jìn)行各種參數(shù)控制,使得細(xì)胞基質(zhì)在適宜的環(huán)境中生長����。
接種的毒種應(yīng)符合菌毒種種子批的管理規(guī)范,包裝用質(zhì)粒應(yīng)符合一定的質(zhì)量管理要求���,復(fù)蘇和擴(kuò)增后的種子或包裝用質(zhì)粒向生產(chǎn)用細(xì)胞基質(zhì)接種時(shí)�����,應(yīng)符合規(guī)定或驗(yàn)證后的擴(kuò)增和接種程序��,還要避免外源污染�����。
細(xì)胞培養(yǎng)過程應(yīng)對溫度范圍�、pH�、CO2、接種活細(xì)胞密度�����、代次等過程工藝參數(shù)實(shí)時(shí)進(jìn)行控制����。以質(zhì)粒轉(zhuǎn)染的AAV上游工藝為例,還需要對細(xì)胞代次�、質(zhì)粒DNA(輔助質(zhì)粒、血清型質(zhì)粒�、目的質(zhì)粒)轉(zhuǎn)染比例����、轉(zhuǎn)染試劑與質(zhì)粒DNA的比例等工藝參數(shù)進(jìn)行評估和工藝研究�。
在生產(chǎn)用容器內(nèi)病毒載體達(dá)到峰值時(shí),通常是收獲的時(shí)刻�����,收獲時(shí)機(jī)是對目標(biāo)物質(zhì)收率和活性等質(zhì)量屬性有重要影響的關(guān)鍵工藝參數(shù)��,可通過縮小模型和單因子法等進(jìn)行試驗(yàn)�����,對收獲時(shí)機(jī)進(jìn)行確認(rèn)����,此時(shí)目標(biāo)物質(zhì)與眾多物質(zhì)混雜在一起,目標(biāo)物質(zhì)定量困難���,通常需要靠工藝驗(yàn)證后的參數(shù)來控制��,這是生產(chǎn)中重要的研究環(huán)節(jié)之一�。
收獲后料液的裂解方式和核酸酶消化方式對于產(chǎn)品的收率和工藝相關(guān)雜質(zhì)殘留的起始水平有重大影響。裂解液的類型和裂解時(shí)間����、核酸酶的用量及消化時(shí)間等需要通過評估并確定風(fēng)險(xiǎn)等級再進(jìn)行工藝探索和驗(yàn)證。收獲后的純化有過濾����、離心����、層析、濃縮和緩沖液置換等步驟�,是逐步去除雜質(zhì)的過程,這一過程帶來的不僅是目標(biāo)物質(zhì)的富集���,同時(shí)還有目標(biāo)物質(zhì)的損失��,最大限度地去除雜質(zhì)和獲得目標(biāo)物質(zhì)最大收率�,是純化的目標(biāo)�。
原液的檢測是工藝要求的重要環(huán)節(jié),經(jīng)過純化的原液需要確定滴度(含量)或活性��,并以此為依據(jù)進(jìn)行半成品配制和成品灌裝�。因此,即使不對原液實(shí)施放行檢驗(yàn)�,也要有中間過程控制檢查和樣品檢測�����。
成品配方配制和制備操作及過程����,也是影響成品穩(wěn)定性的重要因素�。基因治療制品一般都為無菌制品��,需要采用無菌工藝進(jìn)行灌裝�,可以參照國家藥品監(jiān)督管理局發(fā)布的《無菌工藝模擬實(shí)驗(yàn)指南》和《除菌過濾技術(shù)及應(yīng)用指南》實(shí)施。
3�����、生產(chǎn)工藝中間過程的樣品測試和留樣
遵循質(zhì)量源于設(shè)計(jì)(QbD)的原則����,一般在產(chǎn)品開發(fā)初期會確定目標(biāo)產(chǎn)品質(zhì)量概況(QTPP)。QTPP考慮到產(chǎn)品的安全性和有效性�����,在理論上期望產(chǎn)品達(dá)到的一系列質(zhì)量要求,可涉及到劑型����、遞送系統(tǒng)����、包裝系統(tǒng)��,純度��、含量/濃度����、穩(wěn)定性��、安全性���、用藥方式等�。
QTPP描述了產(chǎn)品的設(shè)計(jì)要求��,是確立關(guān)鍵質(zhì)量屬性(CQA)�、關(guān)鍵工藝參數(shù)(CPP)和控制策略(Control Strategy)的基礎(chǔ)。由QTPP導(dǎo)出一系列的產(chǎn)品質(zhì)量屬性�,基于現(xiàn)有知識及已有的藥學(xué)研究、非臨床評價(jià)和臨床評價(jià)數(shù)據(jù)�,通過風(fēng)險(xiǎn)評估確定其對產(chǎn)品的安全性和有效性的影響,識別出關(guān)鍵質(zhì)量屬性���,加上現(xiàn)有對工藝和產(chǎn)品的理解和知識���,形成初步的控制策略��。
在ICH Q10中控制策略的定義是“為了確保工藝性能和產(chǎn)品質(zhì)量����,基于目前對于產(chǎn)品和工藝的理解而形成的一套計(jì)劃設(shè)定的控制措施����,包括生產(chǎn)工藝參數(shù),與原液和制劑生產(chǎn)相關(guān)的原材料�、輔料及包裝材料的參數(shù)和屬性、設(shè)施和設(shè)備運(yùn)行條件����、過程控制�、產(chǎn)品質(zhì)量標(biāo)準(zhǔn)以及與監(jiān)測和控制的分析方法及頻率”??刂撇呗缘男纬墒菑漠a(chǎn)品的理解到工藝的理解的持續(xù)優(yōu)化過程,通過工藝研發(fā)����、工藝表征等過程,采用DoE和不同的風(fēng)險(xiǎn)評估工具�����,識別影響關(guān)鍵產(chǎn)品質(zhì)量屬性的關(guān)鍵物料屬性、關(guān)鍵工藝參數(shù)(CPP)以及其他變因���,組合成一套控制措施�,來實(shí)現(xiàn)產(chǎn)品質(zhì)量的持續(xù)穩(wěn)定受控���。
中間過程控制樣品檢測是在生產(chǎn)的特定步驟實(shí)施采樣并測試��,對生產(chǎn)工藝進(jìn)行及時(shí)監(jiān)控并實(shí)時(shí)反饋生產(chǎn)過程中的某些產(chǎn)品質(zhì)量情況�����,具體項(xiàng)目和監(jiān)控步驟�,可基于對工藝的理解和知識以及對關(guān)鍵質(zhì)量屬性的影響��,經(jīng)過風(fēng)險(xiǎn)評估確定���。通常會考慮各主要生產(chǎn)操作步驟的產(chǎn)物產(chǎn)量和/或主要雜質(zhì)����,用以監(jiān)控該步驟的工藝性能����,以及支原體��、微生物負(fù)載�,內(nèi)毒素等安全性指標(biāo)��,如:對UPB取樣檢測微生物限度����、內(nèi)毒素、支原體以及外源因子等�,在收獲、層析�����、超濾等步驟會采樣檢測產(chǎn)物產(chǎn)量(如基因組滴度等)以及微生物限度和內(nèi)毒素�。通常在臨床試驗(yàn)藥品生產(chǎn)和商業(yè)化生產(chǎn)初期會加強(qiáng)過程監(jiān)控和檢測,隨著生產(chǎn)批次的增多以及生產(chǎn)經(jīng)驗(yàn)的積累可逐步減少某些檢項(xiàng)和頻率�����。
采樣過程應(yīng)不影響正常的生產(chǎn)��,不能因?yàn)椴蓸佣胛廴尽?/span>
留樣可參照GMP 2010版第二百二十五條和“臨床試驗(yàn)藥品附錄”中第三十六條的要求實(shí)施����,若制劑的組分與原液一致、儲存條件一致����,特別是采用原液和制劑生產(chǎn)過程不進(jìn)行凍存的,可考慮直接對制劑成品進(jìn)行留樣�����。
4�、病毒載體類基因治療制品的生產(chǎn)條件
細(xì)胞基質(zhì)復(fù)蘇后需要逐級擴(kuò)增到一定數(shù)量規(guī)模后才能開始生產(chǎn)過程,擴(kuò)增過程應(yīng)盡量選擇與生產(chǎn)相近的環(huán)境條件進(jìn)行���。所有這些步驟的接種量和擴(kuò)增時(shí)間等工藝參數(shù)都需要通過工藝驗(yàn)證數(shù)據(jù)獲得�����,并且應(yīng)規(guī)定下來形成文件����,指導(dǎo)后續(xù)細(xì)胞基質(zhì)準(zhǔn)備過程��。細(xì)胞基質(zhì)生產(chǎn)結(jié)束時(shí)的代次應(yīng)不大于細(xì)胞庫穩(wěn)定性驗(yàn)證中證明細(xì)胞可以穩(wěn)定傳代的最高限定代次��。
病毒種子批的復(fù)蘇與擴(kuò)增也應(yīng)逐級放大,各種參數(shù)需要工藝驗(yàn)證獲得��,形成文件指導(dǎo)后續(xù)種子準(zhǔn)備過程��。種子批的穩(wěn)定性研究��,應(yīng)與種子的最終用途密切相關(guān)�����,若只是輔助生產(chǎn)�����,生產(chǎn)結(jié)束后種子的代次要低于種子批穩(wěn)定性驗(yàn)證中證明種子穩(wěn)定的最高限定代次��;若種子最后需要形成制品����,并且進(jìn)入人體,需要種子批穩(wěn)定性驗(yàn)證��,證明其在人體中使用達(dá)到極限高代次時(shí)仍然穩(wěn)定�。質(zhì)粒擴(kuò)增依賴于細(xì)菌種子批擴(kuò)增,這一過程可能涉及抗生素的有關(guān)要求�,參見《中國藥典》凡例。質(zhì)粒的制備環(huán)境要求與質(zhì)粒的最終用途有關(guān)�,若是裸質(zhì)粒制品,則需要參照GMP實(shí)施��。若是作為起始原材料使用���,應(yīng)符合2020年版《中國藥典》三部中“人用基因治療制品總論”有關(guān)要求���。不同產(chǎn)品的質(zhì)粒生產(chǎn)需額外注意不同質(zhì)粒生產(chǎn)的共線風(fēng)險(xiǎn),避免質(zhì)粒生產(chǎn)的交叉污染風(fēng)險(xiǎn)�。
5、病毒載體類基因治療制品的純化工藝選擇
純化工藝是制品生產(chǎn)的重要環(huán)節(jié)����,是制品中目標(biāo)物質(zhì)富集、純度提升��、去除雜質(zhì)等的關(guān)鍵步驟�����,是提升制品質(zhì)量的有效步驟之一��。純化工藝設(shè)計(jì)與制品的特性密切相關(guān),從總體上講��,分為開放性純化工藝和封閉性純化工藝��。開放性純化工藝的生產(chǎn)管線不能完全封閉����,需要存在與外界直接接觸的過程,如密度梯度離心等��,但這樣的工藝管線有一定的弊端��,既增加污染機(jī)會��,也容易造成泄露����;封閉管線系統(tǒng),相對安全���,在無菌接口的配合下����,基本實(shí)現(xiàn)密閉純化��,此工藝更適用于可進(jìn)行柱純化工藝的制品。
病毒載體基因治療制品顆粒相對較大�,在培養(yǎng)、純化及濃縮過程中可能會因?yàn)閷羟辛γ舾卸Щ?��,因此在進(jìn)行層析及濃縮工藝設(shè)計(jì)時(shí),應(yīng)考慮盡可能地采用減小這些破壞因素的純化工藝��,如控制流速�、減小切向流壓力,但是還必須考慮純化工藝對雜質(zhì)的去除效果���。以AAV的下游層析工藝為例�,需要對層析上樣載量����、流速、上樣pH���、電導(dǎo)率�����、洗脫pH和洗脫柱體積等進(jìn)行工藝探索和驗(yàn)證��。純化工藝的研究和驗(yàn)證����,需要基于質(zhì)量源于設(shè)計(jì)的理念,并且運(yùn)用風(fēng)險(xiǎn)評估和實(shí)驗(yàn)設(shè)計(jì)等工具摸索和驗(yàn)證�,確定工藝步驟和參數(shù)等。純化工藝還有一個(gè)重要的原則需要推薦�,即盡可能地選用容易擴(kuò)大生產(chǎn)規(guī)模的純化工藝方法,為后續(xù)工藝放大和擴(kuò)大生產(chǎn)提前做好準(zhǔn)備�����。
6�����、病毒載體基因治療制品分裝工藝的考慮因素
經(jīng)純化獲得的原液一般需要進(jìn)行相應(yīng)的檢測��,指導(dǎo)后續(xù)半成品配制(若適用)和成品灌裝�,對于從原液到成品為連續(xù)工藝的制品,可在風(fēng)險(xiǎn)評估及工藝驗(yàn)證的基礎(chǔ)上進(jìn)行原液風(fēng)險(xiǎn)放行����。配方溶液配制步驟和操作對于病毒載體基因治療制品也非常重要,經(jīng)工藝驗(yàn)證獲得的方法和參數(shù)應(yīng)固化成生產(chǎn)文件����。
灌裝需要進(jìn)行模擬灌裝驗(yàn)證�����,確認(rèn)灌裝環(huán)境�、極端灌裝條件以及灌裝量等����,這一步驟應(yīng)參考國家藥品監(jiān)督管理局頒布的“無菌工藝模擬實(shí)驗(yàn)指南”和“除菌過濾技術(shù)及應(yīng)用指南”實(shí)施���。
7�、病毒載體類基因治療制品工藝驗(yàn)證的考慮因素
FDA在2011年頒布的《工藝驗(yàn)證:一般原則與實(shí)踐》里將工藝驗(yàn)證定義為收集并評估從工藝設(shè)計(jì)階段一直到商業(yè)化生產(chǎn)的數(shù)據(jù)�����,用這些數(shù)據(jù)建立科學(xué)依據(jù)來證明該工藝能夠始終如一地生產(chǎn)出優(yōu)質(zhì)產(chǎn)品��。它將整個(gè)工藝驗(yàn)證分為3個(gè)階段(工藝設(shè)計(jì)�����、工藝確認(rèn)和持續(xù)工藝確認(rèn))�,整合了ICH Q8�、Q9�、Q10、Q11等相關(guān)指南的一些新理念(如質(zhì)量源于設(shè)計(jì)�、生命周期方法、質(zhì)量風(fēng)險(xiǎn)管理�、藥物質(zhì)量體系等),涵蓋了產(chǎn)品的整個(gè)生命周期����。
第一階段工藝設(shè)計(jì)的主要目標(biāo)是開發(fā)一個(gè)穩(wěn)健的生產(chǎn)工藝以及建立適當(dāng)?shù)目刂撇呗裕源_保產(chǎn)品質(zhì)量始終如一�,然后在第二階段中對控制策略進(jìn)行確認(rèn),將其應(yīng)用于商業(yè)化工藝中��。第一階段的主要工作是定義目標(biāo)產(chǎn)品質(zhì)量概況(QTPP)與評估確定關(guān)鍵質(zhì)量屬性(CQA)��,并進(jìn)行初始的風(fēng)險(xiǎn)評估以對工藝參數(shù)進(jìn)行初步分類����。這個(gè)評估主要是基于已有知識或早期開發(fā)工作,評估結(jié)果為工藝表征研究提供基礎(chǔ)�,從關(guān)鍵性分類中確認(rèn)和縮減了工藝參數(shù)數(shù)目,有助于高效建立參數(shù)范圍����。工藝表征研究可以根據(jù)需要使用單變量與多變量研究進(jìn)行實(shí)驗(yàn)����,使用實(shí)驗(yàn)設(shè)計(jì)(DoE)方式有助于理解多個(gè)參數(shù)的交互作用����,從而幫助最終確定關(guān)鍵工藝參數(shù)(CPP)和非關(guān)鍵工藝參數(shù)(Non-CPP)及其可接受范圍,進(jìn)而支持建立工藝控制策略����。控制策略包含的元素包括但不限于原材料控制��、工藝參數(shù)控制��、中間工藝控制���、性能參數(shù)監(jiān)控、中間品和產(chǎn)品質(zhì)量標(biāo)準(zhǔn)��、穩(wěn)定性考察等��。需要注意的是�����,產(chǎn)品生命周期內(nèi)的風(fēng)險(xiǎn)評估需要根據(jù)相關(guān)分子的先驗(yàn)知識、平臺知識�����,以及來自工藝�����、非臨床與臨床開發(fā)的特定知識和數(shù)據(jù)不斷迭代更新�。
第二階段工藝確認(rèn)的目的是證明該工藝可以按預(yù)期工作并產(chǎn)生可重現(xiàn)性的商業(yè)化產(chǎn)品,主要包括廠房���、公用設(shè)施����、設(shè)備的確認(rèn)(需要在工藝性能確認(rèn)之前完成)和工藝性能確認(rèn)(PPQ)�。監(jiān)測PPQ批次是工藝驗(yàn)證的主要要素,以顯示工藝的一致性�、雜質(zhì)去除能力,以及滿足CPP����、中間工藝控制、工藝性能參數(shù)與CQA的可接受標(biāo)準(zhǔn)的能力����。PPQ證明了在商業(yè)化生產(chǎn)規(guī)模下工藝設(shè)計(jì)的有效性和工藝控制策略的適用性�。
在PPQ研究中�,需要運(yùn)行的批次數(shù)是基于風(fēng)險(xiǎn)來確定的,具有完整的先驗(yàn)知識�����、工藝開發(fā)��、工藝表征和多批成功放大生產(chǎn)(生產(chǎn)規(guī)模同商業(yè)化生產(chǎn))數(shù)據(jù)和經(jīng)驗(yàn)的����,PPQ則需要較少的運(yùn)行批次,反之�,則需要較多的運(yùn)行批次。CDE頒布的《體內(nèi)基因治療產(chǎn)品藥學(xué)研究與評價(jià)技術(shù)指導(dǎo)原則(試行)》中對于生產(chǎn)工藝的確認(rèn)與驗(yàn)證的批次描述到:驗(yàn)證研究的批次數(shù)量與工藝的復(fù)雜性����、變異度����,以及前期工藝研究的充分性、平臺經(jīng)驗(yàn)等有關(guān)�,一般不少于3批����,若有其他特殊情況�,建議提前與監(jiān)管機(jī)構(gòu)開展溝通交流。目前�,很多基因治療產(chǎn)品都是用于罕見病,市場需求量很小���,建議在前期開發(fā)和生產(chǎn)數(shù)據(jù)較多���,以及對產(chǎn)品和工藝?yán)斫獗容^充分的情況下,企業(yè)可基于風(fēng)險(xiǎn)評估�����,提前與CDE進(jìn)行溝通是否酌情減少PPQ的生產(chǎn)批次(做1~3批)�。
第三階段持續(xù)工藝確認(rèn)(CPV)是在產(chǎn)品生命周期內(nèi)將繼續(xù)進(jìn)行工藝?yán)械某掷m(xù)監(jiān)測與評價(jià)。持續(xù)工藝監(jiān)測提供了證據(jù)�,表明該工藝在商業(yè)化生產(chǎn)工藝中仍處于控制狀態(tài)且處于已驗(yàn)證狀態(tài)。
8�、基因治療制品中非目標(biāo)病毒的清除和工藝驗(yàn)證
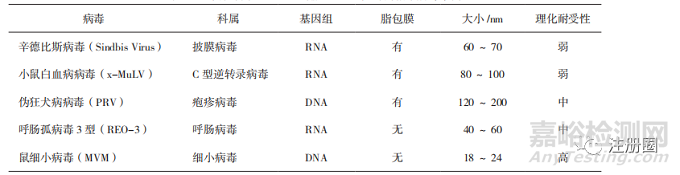
在2022年9月ICH Q5A(R2)征求意見稿中,首次納入了基因工程病毒載體和病毒載體衍生產(chǎn)品�,該指南明確指出了對于適用于某些可進(jìn)行病毒清除而不會對產(chǎn)品產(chǎn)生負(fù)面影響的病毒載體類產(chǎn)品,需要進(jìn)行病毒風(fēng)險(xiǎn)控制和必要的病毒滅活/或清除步驟。如何驗(yàn)證非目標(biāo)病毒清除工藝主要考慮以下3點(diǎn):
(1)病毒清除驗(yàn)證指示病毒的選擇
指示病毒主要分為3類:“相關(guān)”病毒�����、特異性“模型”病毒和非特異性“模型”病毒���。
a.“相關(guān)”病毒是指已被鑒定的病毒或其同種病毒�,其可能會污染細(xì)胞或生產(chǎn)過程中使用的任何其他試劑或材料的病毒����。應(yīng)證明滅活/去除工藝能滅活/去除此種病毒。
b. 特異性“模型”病毒是與“相關(guān)”病毒密切相關(guān)(同種或同屬)�,并與其具有類似理化特性的病毒。若得不到“相關(guān)”病毒�����,或它不太適用于清除病毒工藝評價(jià)研究�,應(yīng)使用特異性“模型”病毒代替。
c. 當(dāng)研究目的是確定生產(chǎn)工藝滅活/去除病毒的總體能力��,即確定方法的可靠性時(shí)�,應(yīng)使用具有不同特性的非特異性“模型”病毒進(jìn)行病毒清除特性研究����。選擇的非特異性“模型”病毒要包括以下特性:具有單鏈和雙鏈的DNA和RNA基因組��、脂包膜和非脂包膜����、大小尺寸����、對物理化學(xué)處理的耐受性。
對于病毒載體類產(chǎn)品���,若生產(chǎn)過程中使用了輔助病毒(桿狀病毒�、HSV���、腺病毒)�,在臨床早期階段就應(yīng)該進(jìn)行病毒清除研究����。在臨床早期應(yīng)至少將該輔助病毒作為指示病毒進(jìn)行研究并在工藝性能確認(rèn)(PPQ)階段增加指示病毒驗(yàn)證。生產(chǎn)工藝應(yīng)能充分地清除輔助病毒��,基于風(fēng)險(xiǎn)對對數(shù)下降因子進(jìn)行評估�����。對于HEK293瞬時(shí)轉(zhuǎn)染工藝平臺,在臨床早期階段通常不要求進(jìn)行病毒清除研究�,建議在PPQ前完成驗(yàn)證。
在生物制品許可申請(BLA)/新藥申請(NDA)階段�,病毒滅活步驟(Low pH或S/D滅活)一般選用2種指示病毒,納濾一般選用2種或4種指示病毒�,層析步驟一般選用4種指示病毒。指示病毒選擇需要考慮的其他問題如下:
a. 盡可能地培養(yǎng)出高滴度的病毒�,并對病毒進(jìn)行純化,以減少對驗(yàn)證試驗(yàn)的干擾��;
b. 應(yīng)有一種有效和可靠的測定方法對要測試的每一道生產(chǎn)工藝中所使用的每種病毒進(jìn)行檢測����;
c. 有些病毒可能會對從事研究的人員造成健康損害,對此應(yīng)加以重視��。
對于使用三質(zhì)粒瞬轉(zhuǎn)���,HEK293細(xì)胞表達(dá)的AAV載體產(chǎn)品�,由于過程中不使用輔助病毒����,可以用表1所列病毒作為指示病毒�。
▲ 表1-三質(zhì)粒轉(zhuǎn)染 HEK293 細(xì)胞 AAV 生產(chǎn)時(shí)的指示病毒舉例
若使用含有內(nèi)源性病毒的昆蟲細(xì)胞(如含有彈狀病毒的 Sf9 細(xì)胞)生產(chǎn)病毒載體產(chǎn)品�,需要研究下游工藝清除彈狀病毒的能力����,結(jié)合風(fēng)險(xiǎn)評估,證明整個(gè)生產(chǎn)工藝能充分去除彈狀病毒���,保證產(chǎn)品的安全����。
(2)病毒清除工藝要求
完整的病毒清除工藝應(yīng)同時(shí)包含病毒滅活(如低pH和S/D滅活)和病毒去除步驟(基于結(jié)合/非結(jié)合機(jī)制如陰離子交換層析�,或基于尺寸排阻機(jī)制如納濾)。一般認(rèn)為���,單個(gè)工藝步驟病毒滴度降低4 log及以上為有效的病毒清除步驟����,1~3 log認(rèn)為是輔助的病毒清除步驟��?�?紤]到檢測和統(tǒng)計(jì)誤差�����,病毒滴度下降較少(如低于1 log)的步驟不計(jì)入總?cè)コ笖?shù)內(nèi)。
病毒清除驗(yàn)證一般使用縮小模型(Scale-down Model)進(jìn)行����,需要考慮縮小模型的代表性和樣品的代表性。至少分別做2次獨(dú)立的病毒清除研究來證實(shí)病毒清除的可重復(fù)性��,一般是使用1批次樣品重復(fù)2次����。
因?yàn)閷游鼋橘|(zhì)的使用次數(shù)可能會影響病毒去除的效果,因此���,若工藝中使用層析工藝作為病毒清除的步驟��,在做病毒清除驗(yàn)證時(shí)���,應(yīng)考慮新舊填料對病毒清除效率的影響。
(3)病毒檢測方法
在病毒清除工藝驗(yàn)證中���,病毒的檢測方法要求高靈敏度����、高特異性以及良好的穩(wěn)定性和重復(fù)性。目前�����,病毒檢測的方法主要包括:噬菌斑檢測法(定量)����、TCID50檢測法(半定量)����、qPCR(定量),其他檢測方法還有電鏡法等��。
9�����、病毒載體類基因治療制品工藝驗(yàn)證中UPB的采樣和檢測要求
UPB被翻譯為未經(jīng)處理原液����,是生產(chǎn)工藝驗(yàn)證中用于病毒安全性驗(yàn)證的采樣。采樣點(diǎn)在生產(chǎn)目標(biāo)物收獲過程的末期���,即在不影響正常生產(chǎn)的情況下����,在最長培養(yǎng)時(shí)間條件(或最有可能采集到目標(biāo)物之外病毒條件)下采集收獲液。
除特殊病毒會與目標(biāo)物質(zhì)一起被濃縮富集外��,其他外源�、內(nèi)源病毒的量在目標(biāo)物質(zhì)達(dá)到最大量值的時(shí)候,通常也達(dá)到峰值�����,因此�,在此階段采樣檢測,更容易測出是否有病毒污染�。
UPB的檢測通常應(yīng)涵蓋工藝驗(yàn)證的連續(xù)多批次,檢測項(xiàng)目以非特異安全檢測項(xiàng)目為主�����,一般包括病毒外源因子檢查��、微生物限度(或無菌檢查)�、支原體檢查、逆轉(zhuǎn)錄酶活性測定等�,若有特殊要求可增加特異性病毒檢查項(xiàng)目或其他檢驗(yàn)項(xiàng)目。
10���、病毒載體基因治療制品生產(chǎn)工藝的穩(wěn)定性研究
制品的質(zhì)量穩(wěn)定或批間一致是制品存在的生命線����,而實(shí)現(xiàn)這一目標(biāo)的關(guān)鍵手段就是生產(chǎn)工藝的穩(wěn)定性。生產(chǎn)工藝的穩(wěn)定性��,包括原輔料持續(xù)來源的穩(wěn)定供給�、細(xì)胞基質(zhì)細(xì)胞庫的穩(wěn)定儲存和使用、菌毒種種子批的穩(wěn)定儲存和使用��、各種生產(chǎn)儀器和設(shè)備的校
準(zhǔn)和使用�、檢驗(yàn)檢測方法的穩(wěn)定和數(shù)據(jù)的可靠性等��。
生產(chǎn)工藝穩(wěn)定性研究通常通過風(fēng)險(xiǎn)評估���,確認(rèn)后進(jìn)行有效措施改進(jìn)���,形成正反饋,進(jìn)而提高工藝穩(wěn)定性�����。
提高生產(chǎn)工藝的穩(wěn)定性是系統(tǒng)而長期的過程��,在生產(chǎn)工藝研發(fā)之初,穩(wěn)定生產(chǎn)工藝需要進(jìn)行大量的工藝表征工作��,以及工藝參數(shù)摸索和確認(rèn)�。在上市申請之前開展工藝驗(yàn)證工作,確定關(guān)鍵工藝參數(shù)及其操作范圍�。
在持續(xù)生產(chǎn)過程中,關(guān)鍵設(shè)備和儀器的更換����,有可能產(chǎn)生影響制品質(zhì)量的風(fēng)險(xiǎn)。為保證生產(chǎn)工藝和制品質(zhì)量可控��,國家藥品監(jiān)管機(jī)構(gòu)通常要求按批準(zhǔn)規(guī)程生產(chǎn)�,不得隨意變動,如需變更���,應(yīng)參照相關(guān)變更程序�。
11�、保證病毒載體類基因治療制品活性的手段
依據(jù)病毒載體基因治療制品的特性,其顆粒相對較大���,對剪切力較敏感���,對紫外線敏感�����,在保存過程容易失活�����。
首先��,工藝方面�,在對產(chǎn)品純化及濃縮時(shí)��,應(yīng)考慮盡可能地采用減小這些破壞因素的工藝��,如控制流速����、減小切向流壓力等�;制劑方面,應(yīng)篩選適宜的處方和內(nèi)包材����,避免病毒產(chǎn)生聚集和吸附,從而導(dǎo)致活性不穩(wěn)定或者下降。
其次�,應(yīng)根據(jù)制品在穩(wěn)定性影響因素方面的特點(diǎn)對制品進(jìn)行穩(wěn)定性測試,以確保制品在適宜的環(huán)境條件下進(jìn)行貯藏和使用��。如使制品盡量處在相對較低溫度以及避免強(qiáng)光照射等�����,從而延長其穩(wěn)定存在時(shí)間�。
凍融對病毒載體活性影響較大,從目前的技術(shù)條件來看�,冷凍到(-70±10)℃以下保存,仍然是較為有效保持病毒活性的方法�;脫離低溫環(huán)境的制品若需再次放回,應(yīng)有凍融穩(wěn)定性研究數(shù)據(jù)支持���。
解凍的制品能在2~8 ℃或室溫的存放時(shí)間,或是否能夠進(jìn)行可控溫度鏈管理���,需要進(jìn)行較為充分的驗(yàn)證����。對解凍后制品的外觀和可見異物的檢查應(yīng)給予足夠重視����。
12��、生產(chǎn)工藝變更原則
基于質(zhì)量源于設(shè)計(jì)(QbD)和全生命周期管理的理念��,為保障基因治療制品臨床前���、臨床試驗(yàn)和上市產(chǎn)品的質(zhì)量一致性要求,生產(chǎn)工藝變更應(yīng)參考國家藥品監(jiān)督管理機(jī)構(gòu)頒布的《藥品上市后變更管理辦法(試行)》和《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》相關(guān)規(guī)定和要求����。涉及重大生產(chǎn)工藝的變更,應(yīng)對變更前后的制品質(zhì)量����、安全性和有效性進(jìn)行比較和評估,以證明變更前后制品特性的一致性��,并確保任何質(zhì)量屬性方面的改變對制品的安全性和有效性無負(fù)面影響。
13�、基因治療制品生產(chǎn)所用質(zhì)粒的生產(chǎn)及穩(wěn)定性考察
由于質(zhì)粒載體(或質(zhì)粒DNA)是重要的起始原材料����,建議納入質(zhì)量管理體系進(jìn)行管理���,視同制品�����,若建立相應(yīng)的主種子批和工作種子批,參照GMP相關(guān)要求生產(chǎn)����,建立質(zhì)量標(biāo)準(zhǔn)并檢驗(yàn)�����。目前����,各種指南沒有強(qiáng)制性條款要求質(zhì)粒應(yīng)按GMP生產(chǎn)。由于基因治療制品的復(fù)雜性和變異性�����,對生產(chǎn)用原材料來源和質(zhì)量有較高的要求���,質(zhì)粒生產(chǎn)工藝和質(zhì)量屬性的穩(wěn)定性影響最終制劑產(chǎn)品的穩(wěn)定性��,基于風(fēng)險(xiǎn)管理要求����,建議生產(chǎn)多批次來考察穩(wěn)定性。
14�����、基因治療制品生產(chǎn)所用質(zhì)粒的灌裝規(guī)格和包材要求
目前��,法規(guī)和指導(dǎo)原則沒有對質(zhì)粒灌裝規(guī)格和包材有明確的要求���,按生產(chǎn)用量分裝,留出檢驗(yàn)檢測和穩(wěn)定性試驗(yàn)的樣品量��。建議穩(wěn)定性試驗(yàn)的樣品包裝材質(zhì)與生產(chǎn)分裝的容器相同�。
15、基因治療制品所用質(zhì)粒的工藝表征和工藝驗(yàn)證研究
建議開展質(zhì)粒的工藝表征和驗(yàn)證研究�,這有利于制品質(zhì)量可控,也有利于監(jiān)管機(jī)構(gòu)評價(jià)產(chǎn)品質(zhì)量�。建議在上游工藝中��,在發(fā)酵罐水平進(jìn)行補(bǔ)料策略、碳源的探索優(yōu)化����,選擇最優(yōu)補(bǔ)料策略及碳源。對發(fā)酵的溶氧����、收獲時(shí)間條件進(jìn)行優(yōu)化,以單位產(chǎn)量及產(chǎn)品DNA同質(zhì)性等為響應(yīng)優(yōu)化工藝參數(shù)��。在下游工藝中����,在裂解工藝進(jìn)行裂解體系、裂解時(shí)間的優(yōu)化�����,優(yōu)化層析工藝的上樣載量和洗脫條件��,以DNA同質(zhì)性比例�����、E.coli宿主DNA殘留��、收率等為響應(yīng)優(yōu)化工藝參數(shù)。
16�����、質(zhì)粒的無菌檢查及取樣要求
基因治療制品生產(chǎn)使用的質(zhì)粒是重要的起始原材料���,基于風(fēng)險(xiǎn)評估和相關(guān)指導(dǎo)原則要求��,按原材料或中間過程控制要求取樣����、留樣��,進(jìn)行無菌檢查����,以保證生產(chǎn)過程(涉及細(xì)胞培養(yǎng))達(dá)到無菌要求。
17��、AAV基因治療制品在放大生產(chǎn)前的潛在風(fēng)險(xiǎn)點(diǎn)
對于AAV基因治療制品生產(chǎn)工藝路線的選擇�,需根據(jù)產(chǎn)品的目標(biāo)質(zhì)量概況進(jìn)行風(fēng)險(xiǎn)評估,測算產(chǎn)品所需的生產(chǎn)規(guī)模和關(guān)鍵質(zhì)量屬性�,包括單位產(chǎn)量、效價(jià)、空實(shí)心率等的要求��,應(yīng)避免在工藝放大后出現(xiàn)重大變更����。
基因組完整性�、rcAAV產(chǎn)生的風(fēng)險(xiǎn)、質(zhì)粒DNA殘留水平����、宿主DNA殘留水平、衣殼錯(cuò)包DNA水平不僅與目的基因(Gene of Interest���,GOI)設(shè)計(jì)有關(guān)���,也和生產(chǎn)使用的細(xì)胞株/上游工藝條件相關(guān),應(yīng)在工藝放大前進(jìn)行工藝可行性評估��,并根據(jù)產(chǎn)品的特性���,結(jié)合實(shí)際情況采用不同分析手段拓展質(zhì)量研究�。
AAV由于其自身特性���,存在易產(chǎn)生聚集以及吸附��、凍融穩(wěn)定性差等問題�,建議在工藝放大前做評估測試,并結(jié)合預(yù)期臨床用途���、劑量規(guī)格����、給藥系統(tǒng)�����、容器密封系統(tǒng)的要求來優(yōu)化篩選制劑處方�����,以提高AAV的制劑穩(wěn)定性��。
18����、AAV基因治療制品的工藝研究
AAV基因治療制品可應(yīng)用實(shí)驗(yàn)設(shè)計(jì)(Design of Experiments,DoE)等工具對關(guān)鍵工藝參數(shù)進(jìn)行開發(fā)��。如在有代表性的縮小規(guī)模的模型測試時(shí),以裂解液滴度�、空實(shí)心比例、宿主DNA殘留等為響應(yīng)進(jìn)行轉(zhuǎn)染條件��,如:活細(xì)胞濃度(Viable Cell Density��,VCD)密度�����、P/D(轉(zhuǎn)染試劑/質(zhì)?�?偭?�,PEI/DNA)��、DNA用量等多變量工藝參數(shù)表征����,得到最優(yōu)轉(zhuǎn)染條件���。對澄清過濾工序的濾器進(jìn)行篩選確認(rèn)并驗(yàn)證�����。對超濾濃縮工序所使用的超濾膜包或膜柱的孔徑�、跨膜壓差、置換次數(shù)等進(jìn)行確認(rèn)并驗(yàn)證�。對層析的載量、洗脫條件等進(jìn)行確認(rèn)����。在除菌過濾工藝環(huán)節(jié)應(yīng)對膜材質(zhì)進(jìn)行篩選確認(rèn)并驗(yàn)證。應(yīng)有穩(wěn)定性數(shù)據(jù)支持灌裝工藝的操作時(shí)長�����,應(yīng)考察AAV制品與包裝材料和給藥器具的相容性�����,增強(qiáng)臨床劑量的準(zhǔn)確度和臨床給藥的安全性����。
七、基因治療制品的質(zhì)量研究及質(zhì)量控制
基因治療制品的質(zhì)量研究應(yīng)貫穿于制品全生命周期�,在質(zhì)量源于設(shè)計(jì)(QbD)的理念下,從生產(chǎn)工藝設(shè)計(jì)(制品特性的研究)����、生產(chǎn)準(zhǔn)備(原輔料����、種子批和細(xì)胞庫的檢定)�����、生產(chǎn)純化(工藝的驗(yàn)證)����、原液成品(放行標(biāo)準(zhǔn)的制定)、效期制定(穩(wěn)定性研究)進(jìn)行質(zhì)量研究����,控制質(zhì)量風(fēng)險(xiǎn)�,確保制品的安全性、有效性和質(zhì)量可控性����。放行檢驗(yàn)研究包括:如何設(shè)立質(zhì)量控制項(xiàng)目、質(zhì)量控制方法的選擇和標(biāo)準(zhǔn)限度范圍等��;方法學(xué)研究及標(biāo)準(zhǔn)物質(zhì)研究�;成品穩(wěn)定性研究及質(zhì)量評價(jià)項(xiàng)目設(shè)立原則等。
1���、基因治療制品的質(zhì)量控制研究與風(fēng)險(xiǎn)評估
風(fēng)險(xiǎn)評估既是一種方法或手段�,也是一種思維方式?�;蛑委熤破返馁|(zhì)量風(fēng)險(xiǎn)評估貫穿于全生命周期質(zhì)量控制體系���,其中的風(fēng)險(xiǎn)識別尤其重要��,因?yàn)檫@是風(fēng)險(xiǎn)評估的開端�。
風(fēng)險(xiǎn)分析是風(fēng)險(xiǎn)評估中的重要環(huán)節(jié)���,基因治療制品的特殊性和復(fù)雜性都對制品質(zhì)量風(fēng)險(xiǎn)分析提出了更高的要求�,這一過程涉及到制品生產(chǎn)工藝的各個(gè)相關(guān)環(huán)節(jié)�����。
在工藝研究中應(yīng)先明確產(chǎn)品目標(biāo)質(zhì)量概況�,利用對制品特性認(rèn)識、前期研發(fā)的經(jīng)驗(yàn)和行業(yè)的先驗(yàn)知識等通過風(fēng)險(xiǎn)評估方法如風(fēng)險(xiǎn)排名����、故障模式影響分析 (Failure Mode and Effects Analysis, FMEA)等適宜的工具進(jìn)行關(guān)鍵質(zhì)量屬性(CQA)和關(guān)鍵工藝參數(shù)(CPP)的評估,并將評估的結(jié)果輸出到工藝研究中�����。在工藝研究中,可結(jié)合實(shí)驗(yàn)設(shè)計(jì)(DoE)工具進(jìn)行工藝開發(fā)驗(yàn)證�。應(yīng)結(jié)合制品特點(diǎn),對于制品工藝特性關(guān)聯(lián)較大的雜質(zhì)進(jìn)行充分研究����。
基于質(zhì)量源于設(shè)計(jì)(QbD)的整體框架有助于建立系統(tǒng)化、以目標(biāo)為導(dǎo)向的制造工藝開發(fā)流程和全面的分析控制策略�。而風(fēng)險(xiǎn)評估作為實(shí)踐的重要方式,最大的好處是利用各種基于風(fēng)險(xiǎn)的方法確保流程保持在適當(dāng)?shù)南拗品秶鷥?nèi)�,并減少開發(fā)工作的隨機(jī)性,有利于將已有先驗(yàn)知識和開發(fā)實(shí)驗(yàn)數(shù)據(jù)有效地結(jié)合��,從而科學(xué)地識別基因治療制品的關(guān)鍵物料屬性(起始原材料及原材料)�、不同階段產(chǎn)品的關(guān)鍵質(zhì)量屬性(中間品����、原液、成品���、穩(wěn)定性研究樣品)���。
2��、基因治療制品的關(guān)鍵質(zhì)量屬性(CQA)的確定
在基因治療藥物的設(shè)計(jì)和生產(chǎn)中�,根據(jù)預(yù)期的目標(biāo)產(chǎn)品質(zhì)量概況���,如預(yù)期適應(yīng)證和臨床用途���、給藥途徑,劑型�����,給藥系統(tǒng)��、規(guī)格等���,建立藥物研發(fā)的設(shè)計(jì)基礎(chǔ)����。根據(jù)已有的先驗(yàn)知識和不斷開展的早期研究�,采用適當(dāng)?shù)娘L(fēng)險(xiǎn)工具如風(fēng)險(xiǎn)排名、故障模式影響分析��,依據(jù)每一步有可能帶來的質(zhì)量風(fēng)險(xiǎn)���,定義重大����、中度和輕度風(fēng)險(xiǎn)并賦值,確定相應(yīng)的關(guān)鍵質(zhì)量屬性并進(jìn)一步評估工藝開發(fā)關(guān)鍵工藝參數(shù)�����,制定工藝及分析質(zhì)控的策略��,以確保產(chǎn)品安全性和工藝穩(wěn)定性�。
對于病毒載體類基因治療制品,影響其安全性和穩(wěn)定性的因素包括但不限于產(chǎn)品相關(guān)雜質(zhì)����、工藝相關(guān)雜質(zhì)、含量的變化�、核酸和蛋白外殼的表征屬性以及一些安全性因素。其中涉及的檢驗(yàn)項(xiàng)目包括野生型/復(fù)制型病毒檢測�����、聚集體比例�����、空實(shí)比�����、宿主DNA殘留量��、殘留宿主DNA片段大小�����、宿主蛋白殘留�����、生產(chǎn)用原材料殘留��、含量��、純度���、鑒別��、裝量/裝量差異��、生物學(xué)活性���、無菌檢查���、支原體檢查、內(nèi)外源病毒檢查�、細(xì)菌內(nèi)毒素檢查、異常毒性檢查�����、水分(若為凍干制劑)�、外觀(若病毒變化會引起顏色、澄清度的改變)�、可見異物、不溶性微粒等�����。
3����、基因治療制品的表征分析方法
基因治療制品的表征分析主要是指采用分析技術(shù)手段對產(chǎn)品序列、結(jié)構(gòu)和理化性質(zhì)等特征的分析檢測和研究���。根據(jù)產(chǎn)品特點(diǎn)�����,病毒載體類基因治療制品的表征分析方法的確定可從核酸水平���、蛋白外殼水平以及完整病毒顆粒整體水平上考慮。在核酸水平�,需要分析重組載體基因組的完整性和均一性,包括對全基因組/關(guān)鍵基因的測序(如NGS等)����、突變位點(diǎn)的PCR鑒別、對基因組的限制性酶切圖譜�����、瓊脂糖電泳����、毛細(xì)管電泳檢測堿基對大小等。在蛋白水平�,可進(jìn)行鑒別試驗(yàn)[如免疫印跡(WB),酶聯(lián)免疫吸附試驗(yàn)(ELISA)等]����、氨基酸序列分析[如液質(zhì)聯(lián)用(LC-MS)等]�����、翻譯后修飾研究(如LC-MS等)���、修飾位點(diǎn)確定(如LC-MS等)、質(zhì)量肽圖分析(如LC-MS等)�、電荷異質(zhì)性分析[如毛細(xì)管等電聚焦(cIEF)、離子色譜(IEC-HPLC)等]���、蛋白分子量測定[如毛細(xì)管電泳(CE)�、LC-MS等]�、異構(gòu)體/聚集體研究[如分子排阻色譜(SEC-HPLC)等]等。在病毒顆粒整體水平上�,則進(jìn)行顆粒屬性分析[如電鏡、分析型超速離心檢測����、動態(tài)光散射法(DLS),差示掃描熒光法(DSF)�、SEC-HPLC、紫外光譜檢測等]��。
4、基因治療制品質(zhì)控項(xiàng)目的設(shè)置
《基因治療產(chǎn)品非臨床研究與評價(jià)技術(shù)指導(dǎo)原則(試行)》規(guī)定“質(zhì)量研究內(nèi)容應(yīng)覆蓋所有可能與產(chǎn)品安全性����、有效性相關(guān)的特性���,一般包括結(jié)構(gòu)���、鑒別、一般理化特性�����、純度��、生物學(xué)活性��、基因轉(zhuǎn)導(dǎo)效率����、雜質(zhì)、基因型�����、表型等,具體研究項(xiàng)目應(yīng)根據(jù)產(chǎn)品類型��、作用機(jī)制�����、原材料和生產(chǎn)工藝決定”���?��!度芰霾《井a(chǎn)品藥學(xué)研究與評價(jià)技術(shù)指導(dǎo)原則(試行)》規(guī)定“質(zhì)量研究項(xiàng)目需全面充分,盡可能涵蓋所有可能與產(chǎn)品安全性�、有效性相關(guān)的質(zhì)量項(xiàng)目,一般包括鑒別和結(jié)構(gòu)分析��,生物學(xué)活性��,含量�����,純度��、雜質(zhì)和污染物����,以及其他特性等”�。具體檢驗(yàn)項(xiàng)目應(yīng)基于產(chǎn)品類型�、生產(chǎn)工藝、質(zhì)量研究�、穩(wěn)定性和風(fēng)險(xiǎn)評估等確定,一般包括以下項(xiàng)目:
(1)鑒別試驗(yàn)���。根據(jù)基因治療制品的情況,應(yīng)在核酸序列水平采用限制性酶切圖譜分析��、PCR�、RT-PCR、核酸序列測定等方法對載體基因���、目的基因��、缺失片段以及其他影響目的基因表達(dá)的重要部分和基因型進(jìn)行鑒定��。還可同時(shí)在蛋白水平采用電泳����、免疫印跡����、免疫中和試驗(yàn)等方法����,對結(jié)構(gòu)蛋白��、表達(dá)產(chǎn)物��、表型特征等進(jìn)行鑒別����。
(2)純度和雜質(zhì)。在適用的情況下�����,可采用HPLC�����、十二烷基硫酸鈉-聚丙烯酰胺凝膠電泳(SDS-PAGE)�、紫外吸收(如A260/A280比值測定)、分析型超速離心法(AUC)等方法評估總純度���。
對于工藝相關(guān)雜質(zhì)的檢測應(yīng)包括宿主細(xì)胞成分和工藝添加物的殘留��。如來自生產(chǎn)細(xì)胞基質(zhì)或細(xì)菌的宿主細(xì)胞蛋白可采用ELISA���、質(zhì)譜等方法進(jìn)行分析���,宿主細(xì)胞DNA總量檢測可采用DNA雜交、Pico-Green�、Q-PCR、數(shù)字PCR等方法����,殘留宿主DNA片段大小檢測可采用Q-PCR和/或毛細(xì)管電泳等方法,風(fēng)險(xiǎn)基因E1A檢測可采用Q-PCR��、數(shù)字PCR等方法��;若在生產(chǎn)中使用了輔助病毒以及質(zhì)粒DNA可采用Q-PCR等方法進(jìn)行檢測��,牛血清�����、核酸酶����、抗生素可采用ELISA進(jìn)行檢測等;若在生產(chǎn)中使用了其他對人體有害的試劑(如有機(jī)溶劑等)�����,以及轉(zhuǎn)染試劑�、親和配基、裂解試劑�����、超速離心介質(zhì)等��,也應(yīng)在制品放行檢驗(yàn)中采用ELISA�、HPLC等方法進(jìn)行檢測。若在生產(chǎn)過程中使用致瘤細(xì)胞系���,總殘留DNA水平應(yīng)嚴(yán)格控制并保持在最低水平����。
某些工藝相關(guān)雜質(zhì)對人體危害較小�,經(jīng)充分驗(yàn)證,證明生產(chǎn)工藝可以將其有效去除�,可不列入常規(guī)放行檢驗(yàn)項(xiàng)目。
產(chǎn)品相關(guān)雜質(zhì)包括:復(fù)制型病毒或野生型病毒,可采用Q-PCR�、噬斑法等方法進(jìn)行檢測;病毒的聚集體�����、裂解物�����、空殼等非功能形式����,可采用HPLC、AUC法等方法進(jìn)行檢測����;共包裝的非目的基因序列(如包裝錯(cuò)誤基因的AAV),可嘗試堿性瓊脂糖電泳�、CE��、AUC����、質(zhì)譜、NGS等方法進(jìn)行檢測。
對于質(zhì)粒應(yīng)控制不同質(zhì)粒形態(tài)的比例���。其中在AAV的生產(chǎn)中����,可采用瓊脂糖凝膠電泳��、毛細(xì)管電泳���、AEX-HPLC等檢測質(zhì)粒超螺旋比例����。
(3)生物學(xué)活性�����。根據(jù)制品具體情況應(yīng)建立至少1個(gè)生物學(xué)活性指標(biāo)��,其屬性反映制品的生理和/或藥理作用模式��?�;钚詼y定通常包括對基因轉(zhuǎn)移效率(感染性/轉(zhuǎn)導(dǎo)效率/傳遞效率)(如TCID50��、Q-PCR等)、目的基因表達(dá)水平(如Western Blot���、ELISA等)����、表達(dá)產(chǎn)物功能或整個(gè)制品的直接活性進(jìn)行評估�。在可能情況下,活性測定應(yīng)在確定基因治療制品的量效關(guān)系基礎(chǔ)上對目的基因或其產(chǎn)物的功能活性進(jìn)行定量測定�。首先應(yīng)考慮開發(fā)體外生物效價(jià)檢測方法,通常是在體外感染��、轉(zhuǎn)染或轉(zhuǎn)導(dǎo)易感細(xì)胞系���,然后進(jìn)行目的基因表達(dá)產(chǎn)物的一些功能測定(如測定酶活性����、細(xì)胞生長的刺激或抑制����、報(bào)告基因法等)。當(dāng)轉(zhuǎn)基因表達(dá)的生物學(xué)功能表現(xiàn)出的活性范圍過寬或僅能產(chǎn)生半定量甚至僅為定性結(jié)果時(shí)��,需使用ELISA或其他具有嚴(yán)格規(guī)定范圍的免疫學(xué)或生物化學(xué)讀數(shù)的方法測定治療序列的表達(dá)水平���,作為補(bǔ)充或替代的活性測定方法(若該方法可獲得廣泛的特性數(shù)據(jù)證明所有表達(dá)的蛋白質(zhì)都具有生物活性���,則可替代生物活性測定方法)。若體外方法不可行�����,應(yīng)考慮在動物離體組織或整個(gè)動物上的檢測方法����,適當(dāng)情況下,可采用轉(zhuǎn)基因動物或移植了人體組織或系統(tǒng)的動物���?���;钚詼y定需要建立相應(yīng)的活性標(biāo)準(zhǔn)物質(zhì)或參比物質(zhì)����,用于計(jì)算被測試制品的相對效價(jià)或作為對照。溶瘤病毒還應(yīng)檢測溶瘤活性(如TCID50法�,即細(xì)胞半數(shù)殺傷時(shí)的病毒MOI值)和選擇性增殖(一般采用噬斑法或Q-PCR法)或選擇性殺傷活性(如TCID50法或Q-PCR法)。
(4)含量�。應(yīng)對病毒總顆粒數(shù)(如ELISA����、A260/A280��、HPLC等)����、感染性滴度/感染性顆粒數(shù)(如TCID50、噬斑法等)�����、基因組DNA/RNA 或質(zhì)粒的拷貝數(shù)(如Q-PCR�、數(shù)字PCR等)進(jìn)行適宜的組合來測定原液和成品的含量,并用參比物質(zhì)/對照物質(zhì)進(jìn)行比較計(jì)算或?qū)φ湛刂?��。在制品為病毒載體等情況下���,還應(yīng)進(jìn)行感染性滴度/顆粒數(shù)(或基因組滴度)比例的測定和控制。
(5)一般安全性試驗(yàn)��。在生產(chǎn)的各個(gè)步驟中��,因環(huán)境因素和原輔料引入會帶來微生物污染的風(fēng)險(xiǎn)���,具體引入的微生物種類根據(jù)制品的具體情況而定�,可開展無菌檢查���、細(xì)菌內(nèi)毒素檢查���、異常毒性檢查、支原體檢查��、分枝桿菌檢查��、外源病毒因子檢測等����。若病毒載體對于安全性試驗(yàn)有影響,可采用中和抗體中和掉制品本身的病毒毒性后�����,再進(jìn)行檢測����。具體檢測方法與其他生物制品一致,可參見2020年版《中國藥典》�。
(6)其他檢測項(xiàng)目�。應(yīng)根據(jù)相關(guān)制品的特性而定�,通常的檢測項(xiàng)目包括外觀(如性狀、顏色)���、可見異物�����、不溶性微粒�����、pH值����、滲透壓摩爾濃度�、裝量、水分和賦形劑�。根據(jù)具體情況或具體劑型檢測其他物理化學(xué)性質(zhì),如平均粒度及分布����、乳光、折射率、平均Zeta 電位�、平均包封率、釋放效應(yīng)等�。具體檢測方法參見2020年版《中國藥典》。
在檢測項(xiàng)目的設(shè)置上����,應(yīng)根據(jù)制品的自身特點(diǎn)�����,分別在制品的生產(chǎn)各階段全面合理地開展�,以充分論證產(chǎn)品的安全性、有效性和工藝的穩(wěn)定性���。
在基因治療的質(zhì)量研究中����,不只包含上述已列出的檢驗(yàn)項(xiàng)目�,還應(yīng)包括穩(wěn)定性檢查,以及根據(jù)制品自身特點(diǎn)增加的相應(yīng)檢項(xiàng)����。在質(zhì)量研究過程中,應(yīng)根據(jù)實(shí)際情況��,盡可能地開發(fā)和采用靈敏度高、專屬性好的方法來驗(yàn)證產(chǎn)品的安全性和有效性���,并用多種方法相互印證���,以不斷增進(jìn)對制品的了解?�;蛑委熤破返馁|(zhì)量研究貫穿該類制品的研發(fā)��、生產(chǎn)和上市使用全生命周期���,是隨著科技和認(rèn)識水平的不斷提升而不斷完善和提高的過程���。
5、檢測項(xiàng)目的分析方法選擇和標(biāo)準(zhǔn)限度制定
質(zhì)量標(biāo)準(zhǔn)是保證產(chǎn)品質(zhì)量穩(wěn)定���、安全有效的質(zhì)量控制策略的重要組成部分���,質(zhì)量控制項(xiàng)目的制定,應(yīng)重點(diǎn)關(guān)注已證明的在產(chǎn)品安全有效方面有意義的結(jié)構(gòu)表征和生物學(xué)活性等方面�����,即關(guān)鍵質(zhì)量屬性(CQA)。根據(jù)CQA來制定質(zhì)控項(xiàng)目����,選擇的質(zhì)控方法應(yīng)切實(shí)反映相應(yīng)特性變化,方法的自動化程度和普及性也應(yīng)作為考量因素����。質(zhì)量控制方法的建立,建議參照ICH Q14和Q2相關(guān)要求開發(fā)并經(jīng)過充分的驗(yàn)證��。定量試驗(yàn)應(yīng)包括方法的線性�、使用范圍��、準(zhǔn)確度�、精密度(包括重復(fù)性和中間精密度)、檢測限��、定量限和耐用性等驗(yàn)證項(xiàng)目���;定性試驗(yàn)應(yīng)包括特異性��、靈敏度�����、耐用性等驗(yàn)證項(xiàng)目�。
質(zhì)量標(biāo)準(zhǔn)的制定以控制最終產(chǎn)品的質(zhì)量和批間一致性為目的,包括檢驗(yàn)項(xiàng)目�、分析方法、標(biāo)準(zhǔn)限度��。制定質(zhì)量標(biāo)準(zhǔn)����,應(yīng)首先確定檢驗(yàn)方法,并在保證制品安全性的前提下���,充分考慮制品有效性和本身特點(diǎn)����,兼顧生產(chǎn)工藝的穩(wěn)定生產(chǎn)范圍��,結(jié)合檢測方法的變異性�,通過多批次產(chǎn)品的測定并進(jìn)行統(tǒng)計(jì)學(xué)分析來制定一個(gè)合理的限度范圍。
6�、病毒載體的滴度和比滴度
病毒載體的滴度按不同層面可以分為總顆粒數(shù)、基因組滴度和感染滴度��。總顆粒數(shù)即病毒顆粒含量��,一般可采用ELISA方法測定��?���;蚪M滴度是病毒基因組拷貝數(shù),通常通過Q-PCR法或數(shù)字PCR法進(jìn)行定量���。感染滴度是具有感染活性的病毒顆粒含量��,對于具有細(xì)胞殺傷能力的病毒����,通常采用細(xì)胞病變法(如PFU或TCID50法)進(jìn)行病毒感染滴度的測定����;對于不具有細(xì)胞殺傷能力的病毒��,雖然不能直接觀察�,但是可通過檢測是否有病毒基因擴(kuò)增,從而間接使用TCID50法進(jìn)行測定����。
比滴度通常以感染滴度與總顆粒數(shù)的比值或感染滴度與病毒基因組滴度的比值來表示�����,其可以反映單位病毒的感染能力����,是表征制品特性的一個(gè)指標(biāo)�。比滴度可間接衡量制品的有效性����、免疫原性,也可直接衡量工藝的穩(wěn)定性�����,因此�����,需要對其加以規(guī)定和控制���。
7���、復(fù)制型病毒或野生型病毒的檢測
對于基因治療制品����,通常為非復(fù)制型病毒載體��,為保證其安全性�,需要對復(fù)制型病毒進(jìn)行控制。目前���,復(fù)制型病毒檢測方法除了針對復(fù)制相關(guān)基因的PCR法,還包括敏感細(xì)胞感染試驗(yàn)法�。敏感細(xì)胞感染試驗(yàn)法是將待測樣本接種于敏感細(xì)胞上進(jìn)行多次傳代以擴(kuò)增潛在的復(fù)制型病毒�,在培養(yǎng)末期
通過觀察病變���、檢測復(fù)制相關(guān)基因等方式判斷樣本中是否有復(fù)制型病毒��。復(fù)制型病毒檢測方法要求具有較高的靈敏度,對方法進(jìn)行建立及驗(yàn)證時(shí)�����,可以采用復(fù)制型病毒作為陽性對照����,同時(shí)���,設(shè)置靈敏度對照,驗(yàn)證方法的最低檢出水平��。對于有安全級別要求的病毒����,應(yīng)考慮在相應(yīng)的生物安全實(shí)驗(yàn)室中進(jìn)行。
對于溶瘤病毒制品�����,其本身就是復(fù)制型或條件復(fù)制型病毒��,通常會改造病毒基因組以降低其對正常細(xì)胞殺傷性或增強(qiáng)腫瘤細(xì)胞靶向性�。在病毒包裝或生產(chǎn)過程中,這些病毒可能會通過重組或回復(fù)突變形成野生型病毒���。為保障產(chǎn)品安全性���,需要控制野生型病毒,可以考慮針對病毒種子與野生型病毒在核酸序列或生長特性方面的差異���,建立適宜的方法對野生型病毒進(jìn)行檢測����。
對于非產(chǎn)品相關(guān)的內(nèi)、外源復(fù)制型病毒的檢查�����,應(yīng)采用以非特異性方法為主�、特異性方法重點(diǎn)補(bǔ)充的檢查原則;對于需要用中和抗體中和后進(jìn)行的檢查����,應(yīng)對可能漏檢的病毒(可能被中和抗體一起中和的病毒)進(jìn)行特異性檢查補(bǔ)充。對于復(fù)制型病毒或野生型病毒的檢查不能放棄對檢測量和檢測靈敏度的追求����,即只承認(rèn)在現(xiàn)有方法下未檢出。為避免誤判��,檢查過程應(yīng)小心謹(jǐn)慎�����,逐步證明在制品中是否有非目標(biāo)“活”病毒存在�����。
8���、病毒載體工藝相關(guān)雜質(zhì)的質(zhì)量控制要求
病毒載體的工藝相關(guān)雜質(zhì)主要包括宿主DNA殘留����、宿主蛋白殘留及其他生產(chǎn)用物料殘留等�����,若生產(chǎn)中使用了質(zhì)?;蜉o助病毒,還應(yīng)考慮質(zhì)粒DNA或輔助病毒殘留����。殘留物質(zhì)越少越好,然而在實(shí)際生產(chǎn)過程中��,殘留物質(zhì)難以完全去除��。對于各種殘留物質(zhì)��,應(yīng)基于風(fēng)險(xiǎn)評估的原則,加以科學(xué)控制�����。
宿主蛋白在人體內(nèi)會引發(fā)免疫反應(yīng)�����,影響產(chǎn)品的安全性和有效性�,應(yīng)采用高靈敏度的分析方法檢測產(chǎn)品的宿主蛋白殘留。在工藝過程中���,應(yīng)測試和分析純化不同階段的宿主蛋白殘留量����,從而評估工藝的清除能力�����,并結(jié)合給藥劑量評估產(chǎn)品中宿主蛋白殘留的可接受標(biāo)準(zhǔn)����。
宿主DNA殘留則有可能引入致瘤性和致感染性的風(fēng)險(xiǎn)。而對于含有已知致癌基因的宿主細(xì)胞����,如SV40和大T抗原基因�、myc以及活化的ras基因等��,則要嚴(yán)格控制此類基因的殘留�。
目前�����,CDE關(guān)于基因治療產(chǎn)品相關(guān)指導(dǎo)原則提出對宿主DNA殘留片段的規(guī)定�����,這有助于進(jìn)一步降低宿主DNA殘留所帶來的致癌性和致感染性風(fēng)險(xiǎn)��。根據(jù)目前的研究顯示�����,最小功能基因的大小約為200 bp����。
9、質(zhì)粒DNA殘留的質(zhì)量控制要點(diǎn)
在AAV的三質(zhì)粒生產(chǎn)中�,質(zhì)粒作為一種生產(chǎn)起始原材料,需要對其殘留予以檢驗(yàn)和控制。制品中質(zhì)粒DNA殘留的質(zhì)量控制���,主要用Q-PCR擴(kuò)增特定區(qū)域檢測���。為簡化檢驗(yàn)步驟,該區(qū)域最好為轉(zhuǎn)染的三質(zhì)粒所共有��;為明確區(qū)分質(zhì)粒DNA��、宿主DNA和重組AAV病毒DNA�����,該區(qū)域不應(yīng)在宿主基因組和重組AAV病毒基因組中出現(xiàn)�����。針對該區(qū)域設(shè)計(jì)特異性引物����,建立Q-PCR或數(shù)字PCR檢測方法,并進(jìn)行驗(yàn)證后����,納入質(zhì)控項(xiàng)目����。
10��、基因治療制品檢測方法驗(yàn)證和可接受標(biāo)準(zhǔn)
基因治療制品不是一個(gè)“純”的單一物質(zhì)���,是包括蛋白、核酸和糖組成的復(fù)雜結(jié)構(gòu)��,而且病毒類基因治療制品是有活力的病毒載體����。出于生物安全性考量,對于活病毒載體的檢驗(yàn)仍須是全面嚴(yán)謹(jǐn)?shù)?�,方法的?yàn)證也須是科學(xué)完整的���。病毒類基因治療制品在檢驗(yàn)方法的建立和驗(yàn)證過程中�,要同時(shí)考慮核酸和蛋白外殼兩方面因素����,在檢測方法的建立和驗(yàn)證過程中,應(yīng)考慮不同因素之間的干擾��,相較于其他的治療類生物制品更為復(fù)雜。
確定可接受標(biāo)準(zhǔn)是一個(gè)必須面對的難題�����,不在于定多少量值��,而在于定的是否科學(xué)合理����。當(dāng)有法規(guī)或行業(yè)標(biāo)準(zhǔn)可以參考時(shí),若符合實(shí)際情況�,可直接套用或借用。但是�����,在沒有任何可參考的情況下��,方法驗(yàn)證中的相關(guān)參數(shù)可以借鑒如重復(fù)性�、定量限(LOQ)等,長期和多批次數(shù)據(jù)積累的回顧性分析數(shù)據(jù)(如95%的可信區(qū)間)也可用作可接受標(biāo)準(zhǔn)����。
11、標(biāo)準(zhǔn)物質(zhì)的使用
在基因治療制品質(zhì)量控制的各個(gè)階段����,標(biāo)準(zhǔn)物質(zhì)可以簡化檢驗(yàn)方式���,減小誤差,是質(zhì)量檢驗(yàn)的一把標(biāo)尺����,可應(yīng)用于質(zhì)量控制的各個(gè)階段。
(1)質(zhì)粒DNA標(biāo)準(zhǔn)物質(zhì)����,主要用于PCR鑒定��、限制性酶切圖譜����。
(2)病毒理化對照物質(zhì),因病毒類基因治療制品的復(fù)雜性����,該類標(biāo)準(zhǔn)物質(zhì)很難覆蓋所有理化項(xiàng)目,可根據(jù)不同的檢驗(yàn)項(xiàng)目制定不同標(biāo)準(zhǔn)物質(zhì)��,如病毒顆粒數(shù)標(biāo)準(zhǔn)物質(zhì)(可用于病毒顆粒數(shù)檢測和蛋白外殼鑒別)���、基因組滴度標(biāo)準(zhǔn)物質(zhì)�、感染滴度標(biāo)準(zhǔn)物質(zhì)、AAV空殼標(biāo)準(zhǔn)物質(zhì)等��。
(3)病毒活性標(biāo)準(zhǔn)物質(zhì)�,對于插入的外源基因,可使用該基因所表達(dá)蛋白的相應(yīng)活性標(biāo)準(zhǔn)物質(zhì)��。溶瘤病毒類則可加入溶瘤活性標(biāo)準(zhǔn)物質(zhì)��。
(4)系統(tǒng)適用性對照品:分離度對照(純度)�����、靈敏度對照(復(fù)制性病毒�、野生型病毒)、陽性對照����、陰性對照等。標(biāo)準(zhǔn)物質(zhì)的制備應(yīng)遵循標(biāo)準(zhǔn)物質(zhì)相關(guān)規(guī)定�����,在其使用領(lǐng)域進(jìn)行充分驗(yàn)證�,包括重復(fù)性����、準(zhǔn)確性���、穩(wěn)定性等����。
12�、標(biāo)準(zhǔn)物質(zhì)的研制
標(biāo)準(zhǔn)物質(zhì)的研制一般包括:①對候選標(biāo)準(zhǔn)物質(zhì)的質(zhì)量檢驗(yàn);②對候選標(biāo)準(zhǔn)物質(zhì)的協(xié)作標(biāo)定�,至少需要3家實(shí)驗(yàn)室;③對候選標(biāo)準(zhǔn)物質(zhì)的穩(wěn)定性研究��。穩(wěn)定性研究包括強(qiáng)制條件試驗(yàn)�����、加速穩(wěn)定性研究�����、長期穩(wěn)定性研究等���。強(qiáng)制條件穩(wěn)定性研究是為考察各種極端因素對產(chǎn)品的影響�,了解影響穩(wěn)定性的因素及可能的降解途徑與降解產(chǎn)物���,為長期穩(wěn)定性研究提供依據(jù)����。包括高溫���、反復(fù)凍融等��,再檢測候選標(biāo)準(zhǔn)物質(zhì)的關(guān)鍵質(zhì)量屬性����,如病毒的外觀��、可見異物�、不溶性微粒、含量��、活性����、無菌等;質(zhì)粒的A260/A280、超螺旋比例��、PCR鑒別�����、限制性酶切圖譜等��。加速穩(wěn)定性通過提高存放溫度探討藥物的穩(wěn)定性����,為長期穩(wěn)定性研究提供依據(jù),具體包括不同溫度下候選標(biāo)準(zhǔn)物質(zhì)的CQA穩(wěn)定性驗(yàn)證��。長期穩(wěn)定性是在實(shí)際儲存條件下開展的穩(wěn)定性研究��,可以作為設(shè)定產(chǎn)品保存條件和有效期的主要依據(jù)��。標(biāo)準(zhǔn)物質(zhì)的制備和標(biāo)定可參考2020年版《中國藥典》三部“生物制品國家標(biāo)準(zhǔn)物質(zhì)制備和標(biāo)定規(guī)程”�。
13、原液和成品質(zhì)量控制策略
原液(DS)和成品(DP)在質(zhì)量控制中�,都是重要的質(zhì)控點(diǎn)�����,其中DP為終點(diǎn)質(zhì)控點(diǎn);DS為分裝前生產(chǎn)過程控制質(zhì)控點(diǎn)���,對分裝定量具有重要意義����。
成品在原液的質(zhì)量控制基礎(chǔ)上�,要考慮由原液到成品這一步所引入的輔料風(fēng)險(xiǎn)和污染風(fēng)險(xiǎn)。在質(zhì)量研究中��,應(yīng)在原液和成品中全面考慮與制品特性�����、安全性���、有效性和工藝穩(wěn)定性相關(guān)的技術(shù)指標(biāo)�����。
DS和DP的檢驗(yàn)共同構(gòu)成了制品的放行檢驗(yàn)�。應(yīng)根據(jù)檢測項(xiàng)目的特性與風(fēng)險(xiǎn)制定相應(yīng)的質(zhì)量控制策略��,將檢測項(xiàng)目分別放在DS和DP階段�����,部分項(xiàng)目若無法在DP或DS中進(jìn)行檢測,或采用其他中間過程控制階段的樣品進(jìn)行檢測更有利于對產(chǎn)品質(zhì)量進(jìn)行控制時(shí)�����,可以考慮通過檢測適當(dāng)?shù)闹虚g產(chǎn)物對產(chǎn)品的質(zhì)量進(jìn)行控制�。
14、基因治療制品的放行檢驗(yàn)
按照2020年版《中國藥典》三部“人用基因治療制品總論”的要求���,基因治療制品的放行檢驗(yàn)主要從結(jié)構(gòu)確證�、有效性��、必要的安全性等方面展開�。選擇切實(shí)可以控制制品質(zhì)量的檢測項(xiàng)目組成放行標(biāo)準(zhǔn),對于不能產(chǎn)生實(shí)際質(zhì)量控制作用的檢測項(xiàng)目���,經(jīng)風(fēng)險(xiǎn)評估后可不納入�。對于設(shè)置檢驗(yàn)項(xiàng)目所在生產(chǎn)流程的具體位置����,可根據(jù)制品特性和工藝特性選擇在適宜的生產(chǎn)過程控制質(zhì)控點(diǎn)。同時(shí)�,還應(yīng)兼顧檢測成本和可及性。
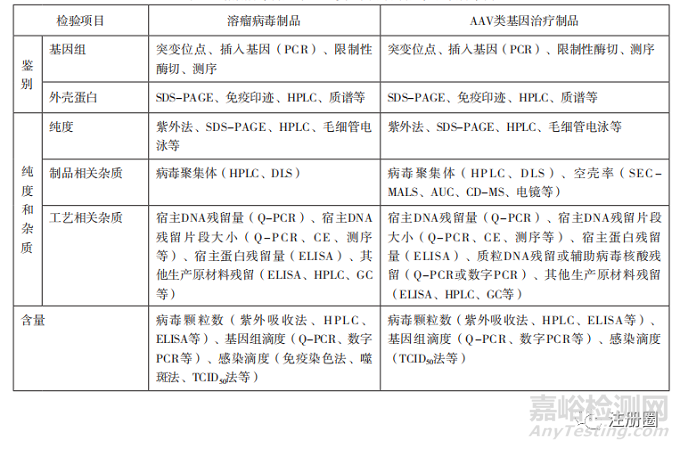
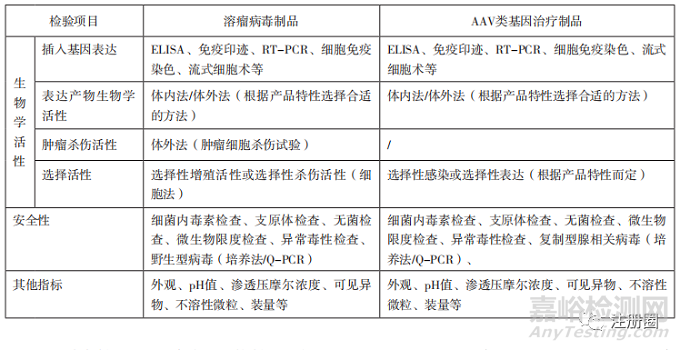
下文選取條件復(fù)制型病毒中有代表性的溶瘤病毒制品�����,非復(fù)制型病毒中有代表性的AAV類基因治療制品�,將建議選擇采用的檢驗(yàn)項(xiàng)目如表2列舉。
表2-溶瘤病毒制品與 AAV 類基因治療制品檢驗(yàn)項(xiàng)目舉例
基因治療制品強(qiáng)調(diào)生產(chǎn)全過程控制��,原液過程控制質(zhì)控點(diǎn)和質(zhì)量標(biāo)準(zhǔn)的設(shè)置����,更有利于生產(chǎn)工藝的平衡和后續(xù)不同規(guī)格的申報(bào)。
15�、AAV基因治療制品病毒空殼率的控制
不同AAV制品的病毒空殼率差異顯著,導(dǎo)致的原因很多�,如產(chǎn)品特性的不同(病毒血清型及插入外源基因差異)、包裝體系的不同(三質(zhì)粒系統(tǒng)/桿狀病毒系統(tǒng))�、純化工藝的不同(超速離心/離子交換層析)、分析方法的不同(AUC/離子交換層析/SEC-MALS)��。目前�,各種指南都建議對AAV基因治療制品的空殼率進(jìn)行控制,但控制范圍���、設(shè)定原則和標(biāo)準(zhǔn)還沒有統(tǒng)一���。當(dāng)前各制品均缺少病毒空殼詳實(shí)的藥效研究���,評價(jià)空殼率與藥效的相關(guān)性,需要基于安全性和有效性的考慮��,綜合考慮產(chǎn)品特點(diǎn)����、臨床試驗(yàn)情況、分析方法變異性等���。
16��、核酸酶殘留限度的考量
在病毒類基因治療制品的生產(chǎn)中����,不管是復(fù)制型病毒載體生產(chǎn)中涉及的宿主DNA�����,還是非復(fù)制型病毒載體生產(chǎn)中所涉及的宿主DNA和質(zhì)粒DNA�����,均需要在工藝中進(jìn)行去除。目前���,生產(chǎn)企業(yè)一般采用核酸酶去除制品中的游離核酸��。這一過程可能會帶來核酸酶殘留,需對其加以控制�。這類工藝相關(guān)雜質(zhì)的可接受水平和標(biāo)準(zhǔn)限度,需要結(jié)合非臨床和/或臨床數(shù)據(jù)或研究經(jīng)驗(yàn)及業(yè)界與監(jiān)管共識等合理制定�。
17、基因治療制品中宿主DNA殘留(HCD)限度
和核酸片段大小的控制各種指南中都建議對宿主細(xì)胞 DNA 殘留量和片段大小進(jìn)行控制�,生產(chǎn)中若使用了腫瘤細(xì)胞系(如 Hela 細(xì)胞),或攜帶致癌基因�、病毒改造序列的細(xì)胞(如 HEK293T),建議盡量將殘留 DNA控制在 10 ng/劑以內(nèi)����,將 DNA 殘留片段大小控制在 200 bp 以下。目前的工藝條件下很難實(shí)現(xiàn)這個(gè)要求����,需要生產(chǎn)企業(yè)不斷改進(jìn)生產(chǎn)工藝提高產(chǎn)品質(zhì)量,同時(shí)開發(fā)出科學(xué)合理的檢測方法���。
八��、基因治療制品的穩(wěn)定性研究
以病毒為載體的基因治療制品具有遺傳不穩(wěn)定性和變異性�,如何保證制品在貨架期的穩(wěn)定存在和在運(yùn)輸時(shí)的惡劣環(huán)境下穩(wěn)定存在以及使用時(shí)對溫度變化耐受等,都需要實(shí)驗(yàn)來模擬回答����。影響穩(wěn)定性的因素可能包括:內(nèi)包材溶出物、儲存時(shí)長����、運(yùn)輸顛簸、高溫���、光照等���。穩(wěn)定性的研究就是對制品在使用期間出現(xiàn)的特殊情況造成的影響給予預(yù)測性提前回答,是制品商業(yè)化的重要保障方案之一���。
1�����、基因治療制品中與穩(wěn)定性相關(guān)的特性
基因治療制品是一類特殊的生物制品����,與蛋白藥物相比,病毒載體類基因治療制品具有自身的特點(diǎn):有效成分是具有核酸和蛋白質(zhì)的生命體��,受環(huán)境條件變化的影響較大���,對熱����、紫外線���、凍融等因素特別敏感,病毒顆粒在保存過程中容易聚集����,劑型大多為液體(因凍干過程對其生物學(xué)活性具有明顯影響)等,應(yīng)根據(jù)制品的特點(diǎn)����,開展相應(yīng)的穩(wěn)定性研究。
2��、基因治療制品穩(wěn)定性研究的主要內(nèi)容
開展穩(wěn)定性研究之前����,需建立穩(wěn)定性研究的整體計(jì)劃或方案���,包括研究樣品、研究條件���、研究項(xiàng)目�����、研究時(shí)間�����、運(yùn)輸研究����、研究結(jié)果分析等方面���。穩(wěn)定性研究一般包括長期穩(wěn)定性研究��、加速穩(wěn)定性研究和強(qiáng)制條件試驗(yàn)研究����。長期穩(wěn)定性研究是在實(shí)際儲存條件下開展的穩(wěn)定性研究,可以作為設(shè)定產(chǎn)品保存條件和有效期的主要依據(jù)�����。加速穩(wěn)定性研究是在高于實(shí)際儲存溫度條件下開展的研究����,對于病毒類制品,建議溫度一般設(shè)置為-20 ℃和4 ℃��,25 ℃和37 ℃為較高溫度����,一般作為高溫條件��。強(qiáng)制條件試驗(yàn)研究是在極端情況下進(jìn)行的穩(wěn)定性研究�,如高溫、光照�、振動、反復(fù)凍融����、氧化等。加速穩(wěn)定性研究和強(qiáng)制條件試驗(yàn)研究可以為長期穩(wěn)定性研究方案提供支持性數(shù)據(jù)�����。
3、基因治療制品穩(wěn)定性研究的樣品要求
基因治療制品穩(wěn)定性研究的樣品通常包括原液和成品����,對因不能連續(xù)操作而需保存一定時(shí)間的中間產(chǎn)物也應(yīng)進(jìn)行相應(yīng)的穩(wěn)定性研究,原液若直接灌裝成成品則無需進(jìn)行原液的長期穩(wěn)定性研究�。穩(wěn)定性研究的樣品批次數(shù)量應(yīng)至少為3批。穩(wěn)定性研究樣品的生產(chǎn)工藝與質(zhì)量應(yīng)一致(即具有代表性)����,研究用成品應(yīng)來自不同批次原液。成品穩(wěn)定性研究應(yīng)采用與實(shí)際貯存相同的包裝容器與密閉系統(tǒng)����;原液或中間產(chǎn)物穩(wěn)定性研究可以采用與實(shí)際應(yīng)用相同的材質(zhì)或材料的容器和密封系統(tǒng)�。對于規(guī)格不同的樣品,穩(wěn)定性數(shù)據(jù)不能互相代替��,最高規(guī)格和最低規(guī)格應(yīng)至少各檢定3批,中間規(guī)格的制品可依據(jù)不同濃度規(guī)格之間的內(nèi)在聯(lián)系及相互支持適當(dāng)減少研究批次����,但是檢測項(xiàng)目必須完整���。
4、基因治療制品穩(wěn)定性研究的考察條件
穩(wěn)定性研究條件應(yīng)充分考慮到今后的貯存��、運(yùn)輸及其使用的整個(gè)過程�����。根據(jù)對各種影響因素(如溫度、濕度��、光照�、反復(fù)凍融、振動、氧化、酸堿等相關(guān)條件)的初步研究結(jié)果����,制定長期��、加速和強(qiáng)制條件試驗(yàn)等穩(wěn)定性研究方案����。
溫度:長期穩(wěn)定性研究的溫度條件應(yīng)與實(shí)際保存條件相一致,原液的溫度可設(shè)置為4 ℃或(-70±10)℃,成品的溫度可設(shè)置為(-70±10) ℃��;病毒載體類制品一般不需要強(qiáng)制降解試驗(yàn)��;加速穩(wěn)定性研究的溫度條件一般介于長期與強(qiáng)制條件試驗(yàn)之間���,通?��?梢苑从钞a(chǎn)品可能短期偏離于實(shí)際保存條件的情況,可設(shè)置4 ℃�����、25 ℃和37 ℃�。
反復(fù)凍融:成品的保存溫度一般為(-70±10)℃,在生產(chǎn)、運(yùn)輸和使用過程中可能會發(fā)生凍融�,應(yīng)驗(yàn)證在多次反復(fù)凍融條件下產(chǎn)品質(zhì)量的變化情況��。
光照:病毒對紫外線敏感����,成品在使用過程中可能受到光線中紫外線的照射��,應(yīng)考察光照對產(chǎn)品質(zhì)量的影響���。
濕度:若能證明包裝容器與密封系統(tǒng)具有良好的密封性能,則不同濕度條件下的穩(wěn)定性研究可以省略;否則�����,需要開展相關(guān)研究。臨床用藥情況:對于一些基因治療制品�,若單次給藥時(shí)間較長的(如靜脈滴注)、使用前需要配制或合并的��,應(yīng)開展相應(yīng)的穩(wěn)定性研究����,以評估實(shí)際使用情況下產(chǎn)品的穩(wěn)定性�����。
5、基因治療制品穩(wěn)定性研究的檢測項(xiàng)目
檢測項(xiàng)目應(yīng)包括產(chǎn)品質(zhì)量變化敏感的�,且反映產(chǎn)品安全性和/或有效性的考查項(xiàng)目���,如效價(jià)、純度、雜質(zhì)和含量等��。對于病毒載體類產(chǎn)品穩(wěn)定性研究可參照《生物制品穩(wěn)定性研究技術(shù)指導(dǎo)原則(試行)》和ICH Q5C要求����,結(jié)合產(chǎn)品特性確定檢測項(xiàng)目��。
(1)活性:活性是穩(wěn)定性研究中的重點(diǎn)研究項(xiàng)目����,活性測定通常包括對基因轉(zhuǎn)移效率(感染性/轉(zhuǎn)導(dǎo)效率/傳遞效率)�����、治療序列表達(dá)的水平、表達(dá)產(chǎn)物的功能或整個(gè)制品的直接活性(如腫瘤細(xì)胞殺傷活性等)的測定��?���;钚詰?yīng)盡可能地采用定量的方法測定����?��;钚詼y定需要采用相應(yīng)的活性標(biāo)準(zhǔn)物質(zhì)或參比物質(zhì)���,使用的參考品應(yīng)該是經(jīng)過標(biāo)準(zhǔn)化的物質(zhì)�,還需要關(guān)注應(yīng)用參考品的一致性和其自身的穩(wěn)定性。
(2)純度和雜質(zhì):可采用HPLC、SDSPAGE����、紫外吸收(如A260/A280比值測定)等方法評估產(chǎn)品的總純度水平。雜質(zhì)包括制品相關(guān)雜質(zhì)和工藝相關(guān)雜質(zhì)���,其中制品相關(guān)雜質(zhì)的含量更能反映產(chǎn)品的穩(wěn)定性��,應(yīng)分析載體降解及聚集情況�。在強(qiáng)制因素研究中經(jīng)過檢測工藝相關(guān)雜質(zhì)若未顯著變化��,可經(jīng)過評估不列入長期穩(wěn)定性考察項(xiàng)�����。降解產(chǎn)物的限度應(yīng)根據(jù)臨床前研究和臨床研究所用各批樣品分析結(jié)果的總體情況來制定。長期穩(wěn)定性研究中,若發(fā)現(xiàn)有新的降解產(chǎn)物出現(xiàn)或者是含量變化超出限度時(shí)�����,建議對其進(jìn)行鑒定,同時(shí)開展安全性與有效性的評估�����。
(3)含量:應(yīng)對總顆粒數(shù)�、感染性滴度或感染性顆粒數(shù)��、基因組DNA/RNA進(jìn)行分析����,還應(yīng)對總顆粒數(shù)或基因組拷貝數(shù)等物理數(shù)量與感染性滴度的比例進(jìn)行測定�。
(4)顆粒特性:病毒載體類基因治療制品的顆粒特征容易受到貯存時(shí)間、溫度��、反復(fù)凍融等因素的影響而發(fā)生變化���,是反映制品穩(wěn)定性的重要指標(biāo)�,應(yīng)進(jìn)行考察,檢測項(xiàng)目包括:可見異物�����、不溶性微粒���、粒度和粒度分布����、折射率�����、聚集體分析等���。
(5)其他:一些檢測項(xiàng)目也是穩(wěn)定性研究
中較為重要的方面����,需要加以關(guān)注����,如外觀、pH值����、無菌檢查等�。在特殊條件下�,選用替代方法需要經(jīng)過充分的驗(yàn)證后使用,如采用容器密封完整性測試替代穩(wěn)定性研究過程中的無菌檢查等���。使用非破壞性容器密封完整性測試方法檢測的樣品�,也可用于其他不受影響的質(zhì)控檢測項(xiàng)目����。添加劑或賦形劑在制劑的效期內(nèi)也可能降解,若初步穩(wěn)定性試驗(yàn)有跡象表明這些物質(zhì)的反應(yīng)或降解對藥品質(zhì)量有不良影響時(shí)�,應(yīng)在穩(wěn)定性研究中加以監(jiān)測。穩(wěn)定性研究中還應(yīng)考慮到包裝容器和密封系統(tǒng)可能對樣品具有潛在的不良影響��,在研究設(shè)計(jì)過程中應(yīng)關(guān)注此方面�,如增加包裝系統(tǒng)密封性檢測。
6����、基因治療制品穩(wěn)定性研究的時(shí)間設(shè)置
對于成品��,若有效期為2年及以上��,則需在第12個(gè)和第24個(gè)月,分別進(jìn)行一次考察項(xiàng)目的檢測����,并在24個(gè)月后,根據(jù)具體穩(wěn)定性研究目的設(shè)置適當(dāng)?shù)臅r(shí)間間隔進(jìn)行考察項(xiàng)目的檢定��。對于有效期為1年以內(nèi)的成品��,則每3個(gè)月進(jìn)行一次考察項(xiàng)目的檢測���。長期穩(wěn)定性研究時(shí)間點(diǎn)設(shè)定的一般原則是����,第1年內(nèi)每隔3個(gè)月檢測1次���,第2年內(nèi)每隔6個(gè)月檢測1次�,第3年開始可以每年檢測1次��。若有效期(保存期)為1年或1年以內(nèi)����,則長期穩(wěn)定性研究應(yīng)為前3個(gè)月每月檢測1次,以后每3個(gè)月1次。在某些特殊情況下�,可靈活調(diào)整檢測時(shí)間,如基于初步穩(wěn)定性研究結(jié)果����,可有針對性地對產(chǎn)品變化劇烈的時(shí)間段進(jìn)行更密集的檢測。原則上���,長期穩(wěn)定性研究應(yīng)盡可能地做到產(chǎn)品不合格為止��。產(chǎn)品有效期的制定應(yīng)根據(jù)長期穩(wěn)定性研究結(jié)果設(shè)定��。強(qiáng)制和加速穩(wěn)定性研究也應(yīng)盡可能地觀察到產(chǎn)品不合格��。
7����、基因治療制品的運(yùn)輸穩(wěn)定性研究
基因治療制品要求冷鏈運(yùn)輸���,對產(chǎn)品的運(yùn)輸過程應(yīng)進(jìn)行相應(yīng)的穩(wěn)定性模擬驗(yàn)證研究�����。穩(wěn)定性研究中需充分考慮運(yùn)輸路線、交通工具、距離����、時(shí)間、條件(溫度����、濕度、振動情況等)�����、產(chǎn)品包裝情況(外包裝��、內(nèi)包裝等)���、產(chǎn)品放置情況和監(jiān)控器情況(溫度監(jiān)控器的數(shù)量��、位置等)等�。穩(wěn)定性研究設(shè)計(jì)時(shí)�,應(yīng)模擬運(yùn)輸時(shí)的最差條件,如運(yùn)輸距離����、振動頻率和幅度及脫離冷鏈等。通過驗(yàn)證研究,應(yīng)確認(rèn)產(chǎn)品在運(yùn)輸過程中處于擬定的保存條件下可以保持產(chǎn)品的穩(wěn)定性�,并評估產(chǎn)品在短暫的脫離擬定保存條件下對產(chǎn)品質(zhì)量的影響,應(yīng)對產(chǎn)品脫離冷鏈的溫度�����、次數(shù)��、總時(shí)間等制定相應(yīng)的要求�����。
8��、基因治療制品穩(wěn)定性研究的結(jié)果分析
穩(wěn)定性研究中應(yīng)建立合理的結(jié)果評判方法和可接受的驗(yàn)收標(biāo)準(zhǔn)����。研究中不同檢測指標(biāo)應(yīng)分別進(jìn)行分析,且應(yīng)對產(chǎn)品進(jìn)行穩(wěn)定性的綜合評估����。同時(shí),開展研究的不同批次的穩(wěn)定性研究結(jié)果應(yīng)該具有較好的一致性���,建議采用統(tǒng)計(jì)學(xué)的方法對批間的一致性進(jìn)行判斷��。同一批產(chǎn)品��,在不同時(shí)間點(diǎn)收集的穩(wěn)定性數(shù)據(jù)應(yīng)進(jìn)行趨勢分析�����,用以判斷降解情況�。驗(yàn)收標(biāo)準(zhǔn)的制定應(yīng)在考慮到方法學(xué)變異的前提下���,參考臨床用研究樣品的檢測值對其進(jìn)行制定或修正�,該標(biāo)準(zhǔn)不能低于產(chǎn)品的質(zhì)量標(biāo)準(zhǔn)���。通過穩(wěn)定性研究結(jié)果的分析和綜合評估���,明確產(chǎn)品的敏感條件、降解途徑���、降解速率等信息����,制定產(chǎn)品的保存條件和有效期(保存期)��。
九、結(jié)束語
本概述匯總了基因治療制品研發(fā)�����、生產(chǎn)和質(zhì)量控制中的常見問題�,以2020年版《中國藥典》、CDE和ICH指導(dǎo)原則為基礎(chǔ)�����,參考其他指南和規(guī)范�����,結(jié)合部分行業(yè)專家的經(jīng)驗(yàn)和意見建議�����,撰寫本考慮概述��,希望給從業(yè)者�����、行業(yè)研發(fā)和質(zhì)量控制人員以啟發(fā)�,共同推動基因治療行業(yè)質(zhì)量控制的進(jìn)步�,促進(jìn)產(chǎn)業(yè)的健康發(fā)展����。
參考文獻(xiàn)
[1] 中華人民共和國藥典:三部[S]. 2020:52-56,凡例�����,5-7��,11-20.
[2] 國家藥品監(jiān)督管理局藥品審評中心. 體內(nèi)基因治療產(chǎn)品非臨床研究與評價(jià)技術(shù)指導(dǎo)原則(試行)(2021年第49號)[EB/OL].(2021-12-03)[2023-04-03].
https://www.cde.org.cn/main/news/viewInfoCommon/41bc557bec23a6ebfb0e148cc989f041.
[3] 國家藥品監(jiān)督管理局藥品審評中心. 溶瘤病毒產(chǎn)品藥學(xué)研究與評價(jià)技術(shù)指導(dǎo)原則(試行)(2023年第2號)[EB/OL].(2023-02-10)[2023-04-03].
https://www.cde.org.cn/main/news/viewInfoCommon/09618d0682fc9161adc0a3f63de486f6.
[4] 國家藥品監(jiān)督管理局藥品審評中心. 預(yù)防用DNA疫苗臨床前研究技術(shù)指導(dǎo)原則[EB/OL].(2003-03-20)[2023-04-03].
https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=bee5a01182004cca5c96f4d171cbdbbb.
[5] 國家藥品監(jiān)督管理局藥品審評中心. 預(yù)防用以病毒為載體的活疫苗制劑的技術(shù)指導(dǎo)原則[EB/OL].(2003-03-20)[2023-04-03].
https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=fb0a8557c244ff9e658407cd299e0ed8.
[6] 國家藥品監(jiān)督管理局. 人基因治療研究和制劑質(zhì)量控制技術(shù)指導(dǎo)原則 [EB/OL].(2003-03-20)[2023-04-03].
https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=65a2d7ba914ad1e0ccd72e6a952b0dc6.
[7] 中華人民共和國衛(wèi)生部. 衛(wèi)生部令第79號 藥品生產(chǎn)質(zhì)量管理規(guī)范[S]. 2011.
[8] FDA. Guidance for Industry, Potency Tests for Cellular and Gene Therapy Products[S]. 2011.
[9] FDA. Guidance for Industry, Preclinical Assessment of Investigational Cellular and GeneTherapy Products[S].2013.
[10] FDA. Guidance for Industry, Manufacturing Considerations for Licensed and Investigational Cellular and Gene Therapy Products During COVID-19 Public Health Emergency[S].2021.
[11] FDA. Draft Guidance for Industry, Human Gene Therapy for Neurodegenerative Diseases[S]. 2021.
[12] FDA. Guidance for Industry and Food and Drug Administration Staff, Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use[S]. 2021.
[13] FDA. Guidance for Industry, Long Term Follow-up After Administration of Human Gene Therapy Products[S]. 2020.
[14] FDA. Guidance for Industry Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs) [S]. 2020.
[15] FDA. Guidance for Industry, Human Gene Therapy for Hemophilia[S]. 2020.
[16] FDA. Guidance for Industry, Human Gene Therapy for RareDiseases[S]. 2020.
[17] FDA. Guidance for Industry, Human Gene Therapy for Retinal Disorders[S]. 2020.
[18] FDA. Guidance for Industry, Testing of Retroviral Vector Based Human Gene Therapy Products for Replication Competent Retrovirus During Product Manufacture and Patient Follow-up[S]. 2020.
[19] FDA. Draft Guidance for Industry, Interpreting Sameness of Gene Therapy Products Under the Orphan Drug Regulations[S]. 2020.
[20] FDA. Guidance for Industry, Evaluation of Devices Used with Regenerative Medicine Advanced Therapies[S]. 2019.
[21] FDA. Guidance for Industry, Expedited Programs for Regenerative Medicine Therapies for Serious Conditions[S].2019.
[22] FDA. Same Surgical Procedure Exception under 21 CFR 1271.15(b): Questions and Answers Regarding the Scope of the Exception[S]. 2017.
[23] FDA. Guidance for Industry, Deviation Reporting for Human Cells, Tissues, and Cellular and Tissue-Based Products Regulated Solely Under Section 361 of the Public Health Service Act and 21 CFR Part 1271[S]. 2017.
[24] FDA. Guidance for Industry, Recommendations for Microbial Vectors Used for Gene Therapy[S]. 2016.
[25] FDA. Guidance for Industry, Design and Analysis of Shedding Studies for Virus or Bacteria-Based Gene Therapy and Oncolytic Products[S]. 2015.
[26] FDA. Guidance for Industry, Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products[S]. 2015.
[27] FDA. CVM GFI #218 Cell-Based Products for Animal Use[S]. 2015.
[28] FDA, Guidance for Industry, Determining the Need for and Content of Environmental Assessments for Gene Therapies,Vectored Vaccines, and Related Recombinant Viral or Microbial Products[S]. 2015.
[29] FDA. Guidance for Industry, BLA for Minimally Manipulated, Unrelated Allogeneic Placental/Umbilical Cord Blood Intended for Hematopoietic and Immunologic Reconstitution in Patients with Disorders Affecting the Hematopoietic System[S]. 2014.
[30] FDA. Guidance for Industry and FDA Staff, IND Applications for Minimally Manipulated, Unrelated Allogeneic Placental/Umbilical Cord Blood Intended for Hematopoietic and Immunologic Reconstitution in Patients with Disorders Affecting the Hematopoietic System[S].2014.
[31] FDA. Guidance for Industry, Preclinical Assessment of Investigational Cellular and Gene Therapy Products[S].2013.
[32] FDA. Guidance for Industry, Preparation of IDEs and INDs for Products Intended to Repair or Replace Knee Cartilage[S]. 2011.
[33] FDA. Guidance for Industry, Clinical Considerations for Therapeutic Cancer Vaccines[S]. 2011.
[34] FDA. Final Guidance for Industry, Potency Tests for Cellular and Gene Therapy Products[S]. 2011.
[35] FDA. Guidance for Industry, Cellular Therapy for Cardiac Disease[S]. 2010.
[36] FDA. Guidance for Industry, Considerations for Allogeneic Pancreatic Islet Cell Products[S]. 2009.
[37] FDA. Guidance for FDA Reviewers and Sponsors, Content and Review of Chemistry, Manufacturing, and Control (CMC) Information for Human Somatic Cell Therapy Investigational New Drug Applications (INDs)[S]. 2008.
[38] FDA. Guidance for Industry, Eligibility Determination for Donors of Human Cells, Tissues, and Cellular and Tissue Based Products[S]. 2007.
[39] FDA. Guidance for Industry, Guidance for Human Somatic Cell Therapy and Gene Therapy[S]. 1998.
[40] EMA. Questions and Answers on Comparability Considerations for Advanced Therapy Medicinal Products (ATMP)[S]. 2019.
[41] EMA. The Overarching Guideline for Human Gene Therapy Medicinal Products is the Guideline on the Wuality, Non clinical and Clinical Aspects of Gene Therapy Medicinal Products[S]. 2014.
[42] EMA. Questions and Answers on Gene Therapy[S]. 2014.
[43] EMA. Reflection Paper on Design Modifications of Gene Therapy Medicinal Products during Development[S]. 2009.
[44] EMA. Reflection Paper on Quality, Non-clinical and Clinical Issues Relating Specifically to Recombinat Adeno associated Viral Vectors[S]. 2007.
[45] EMA. Considerations-Oncolytic Viruses[S]. 2008.
[46] EMA. Guideline on Quality, Non-clinical and Clinical Aspects of Medicinal Products Containing Genetically Modified Cells[S]. 2008.
[47] EMA. Guideline on the Non-clinical Studies Required before First Clinical Use of Gene Therapy Medicinal Products[S]. 2006.
[48] EMA. Guideline on Non-clinical Testing for Inadvertent Germline Transmission of the Gene Transfer Vectors[S].2005.
[49] EMA. Reflection Paper on Management of Clinical Fisks Deriving from Insertional Mutagenesis[S]. 2012.
[50] EMA. Guideline on Follow-up of Patients Administered with Gene Therapy Medicinal Products[S]. 2007.
[51] EMA. Guideline on Safety and Efficacy Follow-up and Risk Management of Advanced Therapy Medicinal Products[S].2008.
[52] EMA. Guideline on the Risk-based Approach According to Annex I, Part IV of Directive 2001/83/EC Applied to Advanced Therapy Medicinal Products[S]. 2013.
[53] USP 40. General Information <1047> Gene Therapy Products[S]. 2017.
[54] ICH Q5A(R2): Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin(Draft)[S]. 2022.
[55] ICH Q5C: Stability Testing of Biotechnological/Biological Products[S]. 1995.
[56] ICH Q5D: Derivation and Characterisation of Cell Substrates Used for Production of Biotechnological/Biological Products[S]. 1997.
[57] ICH Q5E: Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process[S]. 2004.
[58] ICH Q6B: Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products[S]. 1999.
[59] ICH Q8(R2): Pharmaceutical Development[S]. 2008.
[60] ICH Q9: Quality Risk Management[S]. 2005.
[61] ICH Q10: Pharmaceutical Quality System[S]. 2008.
[62] ICH Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management[S]. 2019.
[63] ICH Q2(R2): Validation of Analytical Procedures (Draft)[S].2022.
[64] ICH Q14: Analytical Procedure Development (Draft)[S].2022.
[65] 國家藥品監(jiān)督管理局藥品審評中心. 已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)(2021年第31號)[EB/OL].(2021-06-25)[2023-04-03].
https://amr.hainan.gov.cn/himpa/HICDME/zcfg/yp/202107/t20210702_3004469.html.
[66] 國家藥品監(jiān)督管理局. 藥品上市后變更管理辦法(試行)(2021年第8號)[EB/OL].(2021-01-12)[2023-04-03].
https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20210113142301136.html.
[67] 國家藥典委員會&中國食品藥品國際交流中心. 治療用生物制品病毒污染風(fēng)險(xiǎn)控制要點(diǎn)[EB/OL].(2021-01-15)[2023-04-03].
https://www.ouryao.com/thread-708222-1-1.html.
[68] EMEA. Guideline on Virus Safety Evaluation of Biotechnological Investigational Medicinal Products[S].2009.
[69] 國家食品藥品監(jiān)督管理局. 生物組織提取制品和真核細(xì)胞表達(dá)制品的病毒安全性評價(jià)技術(shù)審評一般原則[EB/OL].(2005-12-18)[2023-04-03].
http://gdcec.gd.gov.cn/publicfiles//business/htmlfiles/rzzx/cmsmedia/image/ypzcsp2/doc7644.pdf.