摘 要 Abstract
由于制藥行業(yè)對(duì)專利保護(hù)具有高度依賴性�����,而新藥研發(fā)的高成本又與公眾的用藥需求相沖突��,為解決這個(gè)問題����,藥品專利鏈接制度應(yīng)運(yùn)而生��。美國(guó)Hatch-Waxman 法案作為該制度的起源����,對(duì)美國(guó)乃至全世界的制藥行業(yè)均具有深遠(yuǎn)的影響�����,其通過仿制藥簡(jiǎn)化申請(qǐng)�����、專利聲明、專利挑戰(zhàn)等制度保障了仿制藥產(chǎn)業(yè)的發(fā)展,同時(shí)也通過專利延長(zhǎng)和試驗(yàn)數(shù)據(jù)保護(hù)等方式支持了原研藥的創(chuàng)新����。
Due to the high dependence of pharmaceutical industry on patent protection and the conflicting nature of high cost of new drug research with public demand for medication, the Pharmaceutical Patent Linkage System has emerged as a solution to solve the increasingly serious issue. The Hatch-Waxman Act in the United States, as the origin of this system, has had far-reaching influence on the pharmaceutical industry both in the United States and the world. It ensures the development of the generic drug through mechanisms such as abbreviated generic drug applications, patent declarations, patent challenges, and so on. At the same time, it also helps support new drug innovation through patent extensions and test data protection.
關(guān)鍵詞 Key words
藥品創(chuàng)新����;仿制藥����;藥品專利鏈接��;Hatch-Waxman 法案
drug innovation; generic drug; Pharmaceutical Patent Linkage; Hatch-Waxman Act
藥品是治療疾病��、挽救生命不可或缺的必需品��。公眾對(duì)藥品的兩項(xiàng)基本需求��,一是要“有好藥”,二是要“吃得起”�����。“有好藥”取決于制藥行業(yè)的技術(shù)創(chuàng)新��,但創(chuàng)新藥(原研藥)研發(fā)具有投資大��、風(fēng)險(xiǎn)大��、難度高�����、周期長(zhǎng)且創(chuàng)新成果易被復(fù)制和模仿的特點(diǎn)�����,需要通過加強(qiáng)專利保護(hù)��、維護(hù)新藥的市場(chǎng)獨(dú)占利益等方式保證創(chuàng)新藥的市場(chǎng)回報(bào)��,即通過經(jīng)濟(jì)刺激鼓勵(lì)藥品的研發(fā)和創(chuàng)新。而創(chuàng)新藥通過專利保護(hù)獲得市場(chǎng)壟斷地位后�����,往往價(jià)格較高�����,令諸多患者望而卻步����,仍然難以獲得治療機(jī)會(huì)��。因此�����,“吃得起”變得至關(guān)重要,需要有更多質(zhì)量��、療效與原研藥一致但價(jià)格卻相對(duì)低廉的仿制藥進(jìn)入市場(chǎng)參與競(jìng)爭(zhēng)����,在法律允許的范圍內(nèi)通過打破原研藥對(duì)市場(chǎng)的壟斷以降低藥品價(jià)格����,從而緩解醫(yī)保負(fù)擔(dān)�����,降低用藥成本��。
由此可見,只有將“有好藥”與“吃得起”二者有機(jī)結(jié)合����,才能實(shí)現(xiàn)藥品的可及性�����。實(shí)現(xiàn)藥品的可及性首先需要平衡原研藥與仿制藥的關(guān)系,而藥品專利鏈接制度在藥品審評(píng)審批過程中對(duì)平衡二者關(guān)系發(fā)揮了重要作用�����。該制度將藥品監(jiān)管機(jī)構(gòu)��、專利機(jī)構(gòu)和法院職能進(jìn)行互動(dòng)銜接��,在藥品審評(píng)審批過程中提前梳理潛在的藥品專利風(fēng)險(xiǎn)����;將仿制藥上市許可與原研藥專利關(guān)系和原研藥專利狀態(tài)相鏈接����,保護(hù)藥品創(chuàng)新的同時(shí)鼓勵(lì)和促進(jìn)仿制藥盡快上市�����,進(jìn)而促進(jìn)整個(gè)醫(yī)藥行業(yè)的良性、有序發(fā)展��。
藥品專利鏈接制度起源于美國(guó)的Hatch-Waxman 法案�����, 即《藥品價(jià)格競(jìng)爭(zhēng)與專利期補(bǔ)償法案》(Drug Price Competition and Patent Term Restoration Act)[1]��,由美國(guó)參議員Orrin Hatch 和眾議員Henry A.Waxman 于1984 年聯(lián)合提出����。法案的主要功能就是平衡原研藥企業(yè)與仿制藥企業(yè)的利益����,在保障原研藥專利權(quán)的同時(shí)�����,通過加快仿制藥上市來鼓勵(lì)藥品價(jià)格競(jìng)爭(zhēng)��。在該法案實(shí)施之前,仿制藥的上市許可程序與原研藥一致��,必須通過一系列的臨床試驗(yàn)來證明自身的安全性和有效性。此外,仿制藥企業(yè)只能在原研藥的專利到期之后����,才能利用原研藥的數(shù)據(jù)和信息從事仿制研究和試驗(yàn),這無疑增加了仿制藥的上市難度,同時(shí)也變相增加了原研藥的市場(chǎng)獨(dú)占期����。而Hatch-Waxman 法案則通過對(duì)仿制藥施行簡(jiǎn)化申請(qǐng)����、Bolar 例外、專利挑戰(zhàn)等制度加快了其上市速度����;同時(shí)也通過專利延長(zhǎng)和試驗(yàn)數(shù)據(jù)保護(hù)等方式加強(qiáng)了對(duì)原研藥的保護(hù)��。
在Hatch-Waxman 法案之后����, 美國(guó)又通過了一系列法案和制度[2-5],最終形成了目前的美國(guó)藥品專利鏈接制度����。當(dāng)前�����,已有加拿大、韓國(guó)��、日本等國(guó)家在參考借鑒美國(guó)藥品專利鏈接制度的基礎(chǔ)上[6]�����,結(jié)合本國(guó)的藥品產(chǎn)業(yè)發(fā)展情況建立了相應(yīng)制度�����。
2017 年,我國(guó)明確提出探索建立藥品專利鏈接制度[7]��。隨著2021 年6 月�����,第四次修改的《中華人民共和國(guó)專利法》的正式實(shí)施,最高人民法院��、國(guó)家藥品監(jiān)督管理局、國(guó)家知識(shí)產(chǎn)權(quán)局陸續(xù)發(fā)布配套制度����,《藥品專利糾紛早期解決機(jī)制實(shí)施辦法(試行)》于2021 年7 月4 日正式發(fā)布�����,中國(guó)藥品專利鏈接制度正式建立��,既借鑒吸納了國(guó)際經(jīng)驗(yàn),也具有相當(dāng)程度的中國(guó)特色。
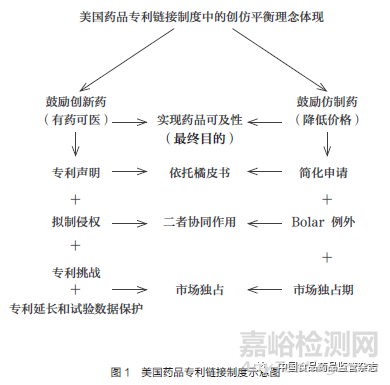
本文將以美國(guó)藥品專利鏈接制度作為切入點(diǎn)�����,通過探析藥品專利鏈接制度的基本結(jié)構(gòu),讓讀者對(duì)該制度形成初步的認(rèn)知和了解。首先����,通過右側(cè)的簡(jiǎn)略圖示(圖1)�����,直觀地感受一下美國(guó)藥品專利鏈接制度的基本架構(gòu)和深植其中的創(chuàng)仿平衡理念��。
一����、橘皮書
橘皮書即《經(jīng)過治療一致性評(píng)價(jià)批準(zhǔn)的藥品》(Approved Drug Products with Therapeutic Equivalence Evaluation)[8]��,因封面為橘色,業(yè)內(nèi)慣稱為“橘皮書”����。橘皮書中收錄的是美國(guó)食品藥品監(jiān)督管理局(FDA)根據(jù)美國(guó)《聯(lián)邦食品藥品和化妝品法案》(Federal Food, Drug, and Cosmetic Act)[9]����,在安全性和有效性基礎(chǔ)上批準(zhǔn)的藥品及其相關(guān)專利與專營(yíng)期信息����。橘皮書制度是藥品專利鏈接制度的基礎(chǔ)����,其核心價(jià)值與功能在于藥品的信息公開。
原研藥企業(yè)在申請(qǐng)新藥上市許可時(shí)��,需要提交與該藥有關(guān)的信息和專利,并通過橘皮書向社會(huì)公布����,橘皮書中收錄的藥品信息和專利將作為仿制藥的參比對(duì)照����,為仿制藥的研發(fā)和申請(qǐng)?zhí)峁┝?ldquo;標(biāo)桿”。在仿制藥申請(qǐng)階段,仿制藥申請(qǐng)人應(yīng)對(duì)照橘皮書中收錄的相關(guān)藥品信息提出不同類型的專利聲明��。同時(shí)��,對(duì)于未列入橘皮書的專利��,原研藥企業(yè)也不得在藥品專利鏈接程序中主張權(quán)利��。
橘皮書于1980 年10 月發(fā)布第一版����,每年會(huì)通過年刊的方式更新過去一年內(nèi)美國(guó)FDA 批準(zhǔn)的藥品及其專利和專營(yíng)期信息��。除此之外��,還會(huì)通過增刊的方式按月更新藥品信息�����。橘皮書現(xiàn)有紙質(zhì)版和網(wǎng)絡(luò)版兩種形式��,便于藥品行業(yè)相關(guān)主體查詢檢索藥品信息��。
二、仿制藥簡(jiǎn)化申請(qǐng)
創(chuàng)新藥的上市����,需要經(jīng)歷候選新藥研發(fā)��、臨床前研究��、臨床試驗(yàn)��、新藥審評(píng)審批與上市后監(jiān)測(cè)等復(fù)雜過程�����,尤其是在新藥申請(qǐng)前需要通過大量的研究試驗(yàn)形成完整����、充分的安全性和有效性數(shù)據(jù)��,整個(gè)過程往往需要耗費(fèi)10 余年的時(shí)間并投入巨額的資金����。而仿制藥的開發(fā)則是建立在原研藥成熟的技術(shù)基礎(chǔ)上[10],仿制藥申請(qǐng)人只需要證明仿制藥與原研藥具有治療等效性即可����。
關(guān)于仿制藥與原研藥的治療等效性����,需要滿足兩項(xiàng)條件,一是藥學(xué)等效性����,即仿制藥與參比制劑(reference listeddrug����,RLD)具有相同的活性成分、劑型����、規(guī)格����、給藥途徑和適應(yīng)癥��;二是生物等效性�����,即在相同的實(shí)驗(yàn)條件下,仿制藥與對(duì)照標(biāo)準(zhǔn)制劑(reference standard�����,RS)在活性成分的吸收程度和速度方面是一致的(差異符合設(shè)定標(biāo)準(zhǔn))[11]����。對(duì)于仿制藥來說����,同時(shí)滿足藥學(xué)等效性和生物等效性條件�����,就可以推定仿制藥與原研藥具有治療等效性����,從而批準(zhǔn)仿制藥的上市許可��。這相比原研藥需要取得完整的安全性和有效性試驗(yàn)數(shù)據(jù)的要求節(jié)省了大量的時(shí)間與經(jīng)濟(jì)成本。
三����、專利聲明
在仿制藥簡(jiǎn)化申請(qǐng)程序中,針對(duì)不同的專利情況��,仿制藥申請(qǐng)人應(yīng)對(duì)照橘皮書向美國(guó)FDA 提出4 種類型的聲明:①橘皮書中沒有收錄相關(guān)專利��;②橘皮書中收錄的相關(guān)專利已過期�����;③承諾在橘皮書中收錄的相關(guān)藥品專利過期前不要求批準(zhǔn)所申請(qǐng)仿制藥上市并注明專利的過期時(shí)間;④橘皮書中收錄的相關(guān)專利是無效的或仿制藥不侵犯相關(guān)專利�����。
原研藥企業(yè)為了保護(hù)自身藥品的市場(chǎng)地位��,一般會(huì)對(duì)其藥品設(shè)立多重的專利保護(hù)��,在新分子實(shí)體或者新化學(xué)實(shí)體專利的基礎(chǔ)上研究制備方法專利�����、用途專利����、劑型專利,組成專利集合并以此成為壟斷市場(chǎng)的“武器”��,對(duì)其他藥企形成專利壁壘��。但是����,在原研藥的眾多專利中,可能只有一部分是真正具有新穎性、創(chuàng)造性和實(shí)用性的產(chǎn)品�����,其余則是為了阻卻競(jìng)爭(zhēng)對(duì)手而刻意設(shè)置的技術(shù)性障礙��,本身不具有科學(xué)價(jià)值�����。而專利聲明制度�����,則給了仿制藥企業(yè)打破原研藥企業(yè)不當(dāng)專利禁錮的機(jī)會(huì)����。此外,由于專利的申報(bào)瑕疵或者橘皮書的更新滯后��,也會(huì)導(dǎo)致部分無效的藥品專利繼續(xù)阻卻仿制藥的上市��。因此����,專利聲明制度設(shè)置了上述4 類聲明內(nèi)容����,仿制藥申請(qǐng)人在進(jìn)行藥品上市許可申請(qǐng)時(shí),需要擇一提交��。對(duì)于提交4 類聲明的仿制藥申請(qǐng)人將產(chǎn)生以下3 種不同的處理結(jié)果��。一是提交第①項(xiàng)和第②項(xiàng)聲明的����,經(jīng)確認(rèn)聲明屬實(shí)并通過技術(shù)審評(píng)后�����,仿制藥即可獲得上市許可����;二是提交第③項(xiàng)聲明的,對(duì)于通過技術(shù)審評(píng)的仿制藥�����,將在原研藥相關(guān)專利到期后方可獲得上市許可�����;三是提交第④項(xiàng)聲明的,仿制藥申請(qǐng)人應(yīng)當(dāng)在提交申請(qǐng)后20 天內(nèi)通知原研藥權(quán)利人(包括藥品專利權(quán)人和上市許可持有人)并對(duì)事實(shí)和法律依據(jù)進(jìn)行說明��,能否獲得上市許可��,除了技術(shù)審評(píng)結(jié)論外�����,還將根據(jù)后續(xù)訴訟結(jié)果處理�����,該程序即為“專利挑戰(zhàn)”程序,也是藥品專利鏈接制度的核心內(nèi)容�����。
四��、專利挑戰(zhàn)
如前文所述�����,在仿制藥申請(qǐng)人提交上述第①至③項(xiàng)聲明時(shí),通過技術(shù)審評(píng)或同時(shí)經(jīng)過專利期限后仿制藥即可獲得上市許可��,這3 種情形屬于仿制藥上市的常規(guī)情形,并不啟動(dòng)專利挑戰(zhàn)程序����。如果仿制藥申請(qǐng)人提交了第④項(xiàng)帶有對(duì)抗性的聲明,則意味著啟動(dòng)了專利挑戰(zhàn)程序��。該程序要求仿制藥申請(qǐng)人應(yīng)當(dāng)在提交含有第④項(xiàng)聲明的申請(qǐng)后20 日內(nèi)通知原研藥的權(quán)利人��,并向其說明相關(guān)專利無效或者不侵犯相關(guān)專利的事實(shí)和法律依據(jù)��,原研藥權(quán)利人可在收到通知后45 天內(nèi)提起專利侵權(quán)訴訟�����。
如果原研藥權(quán)利人提起專利侵權(quán)訴訟����,美國(guó)FDA 將啟動(dòng)30 個(gè)月的批準(zhǔn)等待期(也稱為遏制期或停滯期)����,該期限內(nèi)不停止仿制藥的技術(shù)審評(píng)��,也不批準(zhǔn)仿制藥的上市許可����,用于等待專利侵權(quán)訴訟結(jié)果以確認(rèn)仿制藥與原研藥之間的專利關(guān)系�����。如在批準(zhǔn)等待期內(nèi)法院作出仿制藥侵犯原研藥專利權(quán)的裁決����,仿制藥需要等待原研藥專利到期后才能獲得上市許可;如法院作出不侵犯專利權(quán)的裁決����,通過技術(shù)審評(píng)的仿制藥將獲得上市許可;如法院未能在此期限內(nèi)作出生效裁決����,美國(guó)FDA可對(duì)通過技術(shù)審評(píng)的仿制藥頒發(fā)臨時(shí)上市許可,上市后的侵權(quán)風(fēng)險(xiǎn)由仿制藥申請(qǐng)人自行承擔(dān)��;如仿制藥申請(qǐng)人與原研藥權(quán)利人在此期限內(nèi)達(dá)成和解,美國(guó)FDA 可根據(jù)和解內(nèi)容和技術(shù)審評(píng)結(jié)果作出是否許可上市的決定�����,但和解方應(yīng)將和解協(xié)議提交至聯(lián)邦貿(mào)易委員會(huì)和司法部進(jìn)行反壟斷審查�����,主要目的是避免仿制藥企業(yè)與原研藥企業(yè)通過達(dá)成反向支付協(xié)議等方式損害社會(huì)公眾及其他市場(chǎng)主體利益����;如在此期限內(nèi)原研藥的相關(guān)藥品專利到期����,仿制藥申請(qǐng)人可以將其第④項(xiàng)聲明變更為第②項(xiàng),通過技術(shù)審評(píng)的仿制藥將獲得上市許可����。
如果原研藥權(quán)利人沒有在45 天內(nèi)提起訴訟,美國(guó)FDA 可直接根據(jù)技術(shù)審評(píng)結(jié)果作出是否批準(zhǔn)仿制藥上市的決定�����。需要注意的是��,即使原研藥權(quán)利人沒有在該45 天內(nèi)提起訴訟����,也不妨礙其在仿制藥上市后提起專利侵權(quán)訴訟,原研藥權(quán)利人并不因此喪失其維權(quán)權(quán)利��,其喪失的是30 個(gè)月的遏制期利益��,因?yàn)樵诙糁破趦?nèi)原研藥實(shí)際上仍然享受著市場(chǎng)獨(dú)占利益��。此外��,一旦仿制藥提前上市����,則會(huì)對(duì)原研藥構(gòu)成市場(chǎng)競(jìng)爭(zhēng)��,可能會(huì)導(dǎo)致原研藥價(jià)格大幅下跌����,產(chǎn)生“專利懸崖”現(xiàn)象[12]��,即使通過日后的專利侵權(quán)訴訟也難以挽回其損失,因此絕大部分原研藥權(quán)利人都會(huì)在45 天內(nèi)積極提起訴訟����。
五、市場(chǎng)獨(dú)占期
為了鼓勵(lì)仿制藥盡早上市����,激發(fā)仿制藥企業(yè)挑戰(zhàn)原研藥專利的積極性��,對(duì)于首個(gè)提交第④項(xiàng)聲明進(jìn)行專利挑戰(zhàn)并取得成功的仿制藥(稱之為“首仿藥”)�����,自其上市銷售之日起或者法院作出生效裁決之日起(以較早時(shí)間為準(zhǔn))�����,美國(guó)FDA 將給予其180 天的市場(chǎng)獨(dú)占期����,在此期限內(nèi)美國(guó)FDA 不再批準(zhǔn)其他仿制藥上市��。
該制度對(duì)于仿制藥的研發(fā)與申請(qǐng)具有非常積極的意義,雖然僅規(guī)定了近半年的市場(chǎng)獨(dú)占期�����,但如果首仿藥申請(qǐng)人能夠充分利用市場(chǎng)獨(dú)占期����,除了有助于收回研發(fā)成本外,還能在其他仿制藥大量上市之前確立其市場(chǎng)地位��。
需要注意的是�����,如果仿制藥沒有在以下期限內(nèi)上市�����,將被剝奪市場(chǎng)獨(dú)占期資格�����。①被美國(guó)FDA 批準(zhǔn)上市許可后75 天內(nèi)����;②自提交申請(qǐng)后30 個(gè)月內(nèi)����;③法院判決��、雙方和解或原研藥專利被撤銷后75 天內(nèi)��。具體截止日期的計(jì)算方式為:先在①����、②項(xiàng)之間取時(shí)間較早者,再在時(shí)間較早者與③之間取時(shí)間較晚者��。市場(chǎng)獨(dú)占期制度的目的在于鼓勵(lì)仿制藥上市��,如果仿制藥不把握時(shí)機(jī)盡早上市��,自然也就失去了給予其獨(dú)占地位的意義�����。并且如前文所述�����,首仿藥申請(qǐng)人如果與原研藥權(quán)利人達(dá)成反向支付協(xié)議��,其獲得市場(chǎng)獨(dú)占期后會(huì)阻卻其他仿制藥上市�����,因此有必要對(duì)其施加這種失效限制�����。
六��、Bolar 例外
為了實(shí)現(xiàn)仿制藥的盡早上市��,仿制藥企業(yè)往往需要在原研藥的專利到期前就啟動(dòng)仿制研發(fā)工作��,因?yàn)殚_展藥學(xué)等效性和生物等效性試驗(yàn)等工作都需要使用原研藥的相關(guān)專利��,這種使用方式雖然在仿制藥上市前并不會(huì)對(duì)原研藥的市場(chǎng)份額造成實(shí)質(zhì)性影響��,但顯然是帶有其商業(yè)目的����。依據(jù)美國(guó)的專利法,不能認(rèn)定該涉及原研藥專利的使用行為屬于不視為侵權(quán)的合理使用范疇�����。相反,如果要求仿制藥只能在原研藥專利到期后再開展仿制研究����,在經(jīng)過相對(duì)較長(zhǎng)的研發(fā)試驗(yàn)和審評(píng)審批期間后仿制藥才能實(shí)際上市,這無疑變相延長(zhǎng)了原研藥的專利保護(hù)期�����。因此�����,需要在立法層面明確這種使用原研藥專利研制仿制藥的行為不屬于侵權(quán)行為��,由此就誕生了Bolar 例外制度[13]�����。
簡(jiǎn)言之�����,Bolar 例外就是為了豁免仿制藥開發(fā)過程中的專利侵權(quán)風(fēng)險(xiǎn)����,通過制度明確在原研藥專利到期前,仿制藥企業(yè)及相關(guān)主體為了向美國(guó)FDA 提交仿制藥申請(qǐng)資料而進(jìn)口�����、制造�����、使用專利藥品進(jìn)行試驗(yàn)�����,以獲取相關(guān)數(shù)據(jù)信息的行為����,即使落入了原研藥的專利保護(hù)范圍也不構(gòu)成侵權(quán)。該制度起源于美國(guó)Bolar 公司的一起仿制藥研究行為[14]��,屬于專利權(quán)保護(hù)的例外情形�����,因此名為“Bolar 例外”��。
Bolar 例外制度平衡了原研藥的專利保護(hù)需求和仿制藥的盡快上市需求,縮短了仿制藥的上市遲延�����,使公眾可以在原研藥專利到期后��,盡早獲得價(jià)格相對(duì)低廉的仿制藥��。
七�����、擬制侵權(quán)
對(duì)于仿制藥企業(yè)����,其需要在仿制藥上市之前厘清專利風(fēng)險(xiǎn),否則不但其仿制藥可能面臨退市風(fēng)險(xiǎn)��,還有可能承擔(dān)高額的侵權(quán)賠償����。對(duì)于原研藥企業(yè),前文中也曾提到����,仿制藥一旦上市,原研藥可能面臨“專利懸崖”效應(yīng)�����,即使通過日后的專利侵權(quán)訴訟也難以挽回其市場(chǎng)損失����。因此,為了前置性地解決仿制藥的專利侵權(quán)問題����,使可能發(fā)生的專利侵權(quán)風(fēng)險(xiǎn)在仿制藥上市前得以解決,以此給予原研藥和仿制藥更多的市場(chǎng)確定性����,美國(guó)藥品專利鏈接制度創(chuàng)設(shè)性地運(yùn)用了擬制侵權(quán)制度,即任何人在他人的藥品專利保護(hù)期內(nèi)向美國(guó)FDA 提出仿制藥注冊(cè)申請(qǐng)的����,均構(gòu)成侵犯專利權(quán)。這使得仿制藥的注冊(cè)申請(qǐng)行為具有可訴性��,為專利挑戰(zhàn)中的法院訴訟環(huán)節(jié)提供了制度基礎(chǔ)�����,原研藥專利權(quán)人通過提前訴訟能及時(shí)制止未來可能發(fā)生的侵權(quán)行為,而暫不考慮實(shí)際侵權(quán)和損害情形��。
需要說明的是����,這種人為賦予原研藥專利權(quán)人的訴訟權(quán)利僅是一種程序性權(quán)利,其不會(huì)因此訴訟而獲得實(shí)質(zhì)性賠償����,因?yàn)榉轮扑幧形瓷鲜校瑢?shí)質(zhì)性損害并沒有產(chǎn)生����,如果確認(rèn)仿制藥侵犯了原研藥的專利權(quán),原研藥專利權(quán)人能夠獲得的僅是一種程序性禁令��,即在原研藥專利保護(hù)期滿前仿制藥不得獲批上市����。
從表面上來看,Bolar 例外與擬制侵權(quán)是彼此矛盾的�����,一個(gè)規(guī)定不視為侵權(quán),而另一個(gè)規(guī)定構(gòu)成侵權(quán)����,實(shí)際上正是這兩種制度的有機(jī)結(jié)合才使得仿制藥的研發(fā)與申請(qǐng)能夠有效推進(jìn)����,同時(shí)還能提前規(guī)避專利侵權(quán)風(fēng)險(xiǎn)。在仿制藥研發(fā)階段�����,通過Bolar 例外對(duì)其研發(fā)行為進(jìn)行賦權(quán)����;在仿制藥申請(qǐng)階段,通過擬制侵權(quán)讓原研藥專利權(quán)人對(duì)仿制藥的申請(qǐng)上市行為進(jìn)行訴訟以提前排除專利侵權(quán)風(fēng)險(xiǎn)����。我們也可以這樣理解,僅在申請(qǐng)注冊(cè)的仿制藥技術(shù)本身不存在專利侵權(quán)情形的前提下����,為了開發(fā)仿制藥而使用原研藥相關(guān)專利的行為才不構(gòu)成侵權(quán)。
八����、專利延長(zhǎng)
前幾項(xiàng)制度更多的是為了促進(jìn)仿制藥盡早上市����,為了達(dá)到“創(chuàng)仿平衡”的效果��,對(duì)原研藥因臨床試驗(yàn)和審評(píng)審批延誤的上市時(shí)間����,在專利期限屆滿后給予其適當(dāng)延長(zhǎng),也有利于鼓勵(lì)原研藥企業(yè)對(duì)于新藥研發(fā)的積極性����。在新藥上市過程中,很多專利往往在新藥研發(fā)階段就需要立即申請(qǐng)專利保護(hù)��,而此后的臨床試驗(yàn)和新藥審評(píng)審批環(huán)節(jié)往往又要耗費(fèi)大量的時(shí)間����,很多新藥在上市時(shí)其相關(guān)專利的保護(hù)期已所剩無幾,因此有必要對(duì)其進(jìn)行額外的延長(zhǎng)保護(hù)以保證其市場(chǎng)回報(bào)��。
專利延長(zhǎng)需要由專利權(quán)人在美國(guó)FDA 批準(zhǔn)上市許可后60 天內(nèi)向美國(guó)專利及商標(biāo)局提出申請(qǐng)�����,專利延長(zhǎng)的適用范圍包括產(chǎn)品專利、制備方法專利和醫(yī)藥用途專利�����,具體延長(zhǎng)時(shí)間為臨床試驗(yàn)時(shí)間的一半與審評(píng)時(shí)間之和��,最長(zhǎng)為5 年��,但從藥品上市之日起算專利保護(hù)的總期限不超過14 年����。為了避免專利延長(zhǎng)制度的濫用����,同時(shí)還規(guī)定對(duì)于同一種藥品包含多個(gè)專利的,只能選擇申請(qǐng)其中1 項(xiàng)專利給予延長(zhǎng)����,而且對(duì)于專利權(quán)人沒有盡到合理注意義務(wù)所導(dǎo)致的期限延誤不予保護(hù)。
九����、試驗(yàn)數(shù)據(jù)保護(hù)期
與藥品專利延長(zhǎng)制度相同,藥品試驗(yàn)數(shù)據(jù)保護(hù)期也是給予原研藥權(quán)利人的一項(xiàng)激勵(lì)措施����。試驗(yàn)數(shù)據(jù)是原研藥企業(yè)在藥品研發(fā)過程中為了進(jìn)行新藥的開發(fā)以及證明新藥安全性和有效性所形成的一系列研究數(shù)據(jù)��,主要包括臨床前試驗(yàn)數(shù)據(jù)和臨床試驗(yàn)數(shù)據(jù)��。試驗(yàn)數(shù)據(jù)保護(hù)是區(qū)別于傳統(tǒng)的知識(shí)產(chǎn)權(quán)保護(hù)����,也有別于一般性商業(yè)秘密保護(hù)的一種特殊保護(hù)制度�����,其通過給予試驗(yàn)數(shù)據(jù)權(quán)利人一定期限的數(shù)據(jù)獨(dú)占權(quán)��,來阻止仿制藥申請(qǐng)人使用其數(shù)據(jù)獲得上市許可��,同時(shí)也禁止美國(guó)FDA 運(yùn)用試驗(yàn)數(shù)據(jù)對(duì)仿制藥進(jìn)行審評(píng)審批[15]����。
即使原研藥的專利已經(jīng)到期,在試驗(yàn)數(shù)據(jù)保護(hù)期內(nèi)����,仿制藥申請(qǐng)人也不得利用其試驗(yàn)數(shù)據(jù)進(jìn)行仿制藥申請(qǐng),而仿制藥申請(qǐng)人自行取得試驗(yàn)數(shù)據(jù)的情形除外����。由此可見����,試驗(yàn)數(shù)據(jù)保護(hù)是獨(dú)立于藥品專利保護(hù)的另一種保護(hù)制度�����,其目的也是為了在一定期限內(nèi)限制仿制藥����、me-too 藥和me-better 藥的搭便車行為��,鼓勵(lì)原研藥的開發(fā)�����。
享受試驗(yàn)數(shù)據(jù)保護(hù)的前提是該試驗(yàn)數(shù)據(jù)所支持的藥品已經(jīng)獲得上市許可且試驗(yàn)數(shù)據(jù)本身尚未被披露�����。具體的數(shù)據(jù)保護(hù)期從該藥品獲得美國(guó)FDA 批準(zhǔn)之日起計(jì)算��,其中含有新化學(xué)實(shí)體的新藥享有5 年的數(shù)據(jù)保護(hù)期�����;罕見病藥享有7 年的數(shù)據(jù)保護(hù)期;具有新適應(yīng)癥�����、新劑型或新給藥途徑的已獲批藥品享有3年的數(shù)據(jù)保護(hù)期��;兒科用藥在前述藥品的原保護(hù)期基礎(chǔ)上增加半年����;抗生素享有10 年的數(shù)據(jù)保護(hù)期;新生物制品享有12 年的數(shù)據(jù)保護(hù)期�����。
以上為美國(guó)藥品專利鏈接制度的主要構(gòu)成��,其通過Bolar 例外����、仿制藥簡(jiǎn)化申請(qǐng)、首仿藥180 天市場(chǎng)獨(dú)占期等制度促進(jìn)了仿制藥的上市�����,同時(shí)還通過延長(zhǎng)專利保護(hù)期和試驗(yàn)數(shù)據(jù)保護(hù)制度提高了原研藥企業(yè)的收益預(yù)期。藥品專利鏈接制度的核心價(jià)值在于通過制度設(shè)計(jì)實(shí)現(xiàn)原研藥企業(yè)與仿制藥企業(yè)之間的利益平衡��,既鼓勵(lì)新藥研發(fā)�����,也促進(jìn)仿制藥盡早上市����,構(gòu)建創(chuàng)仿平衡的藥品市場(chǎng)格局,利用原研藥企業(yè)和仿制藥企業(yè)追逐自身經(jīng)濟(jì)效益的動(dòng)能來滿足社會(huì)公眾的用藥需求�����,以此實(shí)現(xiàn)藥品的真正可及��。