在藥企環(huán)境中實(shí)施醫(yī)療器械QMS體系需要認(rèn)真考量及規(guī)劃���,才能第一次就把事情做對(duì),避免任何延誤或額外成本��。
因此��,對(duì)現(xiàn)有的過(guò)程�����、能力和體系進(jìn)行完整評(píng)估很關(guān)鍵,借此可以識(shí)別任何差距��,以及能夠用來(lái)建立醫(yī)療器械QMS體系的資源�����。由藥品Q(chēng)MS體系延伸至醫(yī)療器械QMS的建立��,優(yōu)勢(shì)在于能夠利用現(xiàn)有的過(guò)程和資源���。通過(guò)獨(dú)立認(rèn)證機(jī)構(gòu)獲得ISO 13485質(zhì)量管理體系認(rèn)證能夠加速、協(xié)調(diào)MDR審核過(guò)程���,無(wú)需向公告機(jī)構(gòu)展示所有的產(chǎn)品設(shè)計(jì)或文件資料�����。
完整評(píng)估需囊括企業(yè)內(nèi)部能力��,確保企業(yè)具備與管理和維護(hù)QMS體系對(duì)口的專(zhuān)業(yè)知識(shí)��。至于醫(yī)療器械的研發(fā)和上市后監(jiān)督���,企業(yè)需要配備具有生物技術(shù)���、生物醫(yī)藥、工程���、軟件等相關(guān)背景的專(zhuān)業(yè)人才���。換句話說(shuō),一家提供組合產(chǎn)品的企業(yè)需要保證雇員的組成囊括所有必要的專(zhuān)業(yè)領(lǐng)域��,從而同時(shí)覆蓋藥品和器械��,持續(xù)更新QMS體系�����。
如何設(shè)計(jì)策略���,實(shí)施醫(yī)療器械QMS�����?
制藥企業(yè)須先建立一個(gè)多職能項(xiàng)目團(tuán)隊(duì)���,包含必要的醫(yī)療器械專(zhuān)家�����,來(lái)處理不同方面的事項(xiàng)��。在很多情況下���,企業(yè)會(huì)通過(guò)第三方機(jī)構(gòu)來(lái)進(jìn)行評(píng)估��,制定實(shí)施計(jì)劃���。
考慮到藥品Q(chēng)MS的文件層級(jí)���,高級(jí)別程序文件,比如制度文件���,通常能夠同時(shí)用于器械和藥物�����,因?yàn)檫@些文件通常不包含過(guò)程細(xì)節(jié)以及執(zhí)行階段的角色和職責(zé)��。比如糾正及預(yù)防措施(CAPA)以及投訴管理過(guò)程�����,制度的要素對(duì)于藥物和器械而言是相似的���,而具體執(zhí)行則涉及不同的專(zhuān)業(yè)知識(shí)和體系�����。
部分過(guò)程需要提升�����,確保能夠健全地適用于藥品和醫(yī)療器械�����。例如��,風(fēng)險(xiǎn)管理過(guò)程可能需要不同的專(zhuān)業(yè)知識(shí)和風(fēng)險(xiǎn)分析方法論��,來(lái)處理產(chǎn)品的機(jī)械部分�����。藥品的風(fēng)險(xiǎn)分析主要關(guān)注人體內(nèi)的分子和化學(xué)相互作用�����,與副作用相關(guān)���。醫(yī)療器械的風(fēng)險(xiǎn)分析則關(guān)注器械的設(shè)計(jì)和用戶及患者對(duì)器械的使用��,與意外事件有關(guān)���,因?yàn)獒t(yī)療器械通常涉及更加復(fù)雜的系統(tǒng)。然而��,兩者的高級(jí)別過(guò)程是相似的�����,風(fēng)險(xiǎn)管理團(tuán)隊(duì)會(huì)評(píng)估產(chǎn)品��、對(duì)潛在風(fēng)險(xiǎn)進(jìn)行評(píng)分并設(shè)計(jì)解決方案���,盡量減少對(duì)患者的影響。
高級(jí)別的程序文件和部分過(guò)程可用于不同類(lèi)型的產(chǎn)品�����,而一些低級(jí)別的過(guò)程可能更容易分開(kāi)管理。例如投訴管理過(guò)程��,如果有分別對(duì)應(yīng)藥品和醫(yī)療器械的規(guī)程或工作指引��,投訴管理將會(huì)更加容易��。這與收到的投訴類(lèi)型有關(guān)�����。對(duì)于藥品��,大多數(shù)的投訴與不良事件和副作用有關(guān)�����。對(duì)于醫(yī)療器械�����,收到的大多數(shù)投訴通常是質(zhì)量投訴��,在報(bào)告的時(shí)間點(diǎn)不一定對(duì)患者有影響���。因此���,投訴的評(píng)估需要不同的專(zhuān)業(yè)知識(shí)���,涉及不同的過(guò)程。醫(yī)療器械通常需要對(duì)器械的設(shè)計(jì)和操作進(jìn)行技術(shù)評(píng)估��,而藥品投訴的評(píng)估則需要醫(yī)學(xué)/臨床知識(shí)��。所以說(shuō)��,配備合適的專(zhuān)業(yè)人員���,確保進(jìn)行了適當(dāng)?shù)恼{(diào)查,是至關(guān)重要的�����。
法規(guī)專(zhuān)家角色的區(qū)別
醫(yī)療器械QMS與藥品Q(chēng)MS的要求有所重疊���,但也存在一些明顯的不同之處。我們選擇其中一項(xiàng)要求(法規(guī)專(zhuān)家的角色)作為示例��,分析其中的共性和區(qū)別。
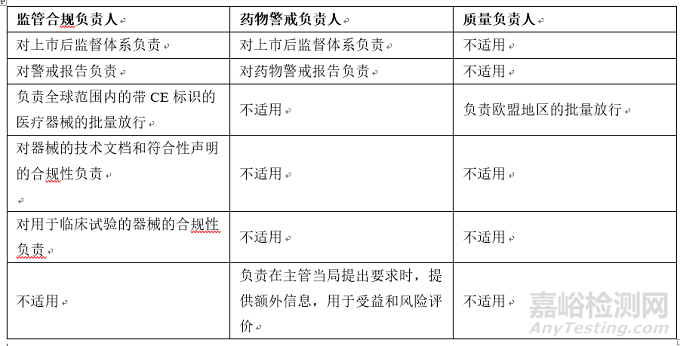
醫(yī)療器械和藥品的QMS體系都要求配備質(zhì)量管理代表���,對(duì)QMS的有效性負(fù)責(zé)���,法規(guī)也對(duì)這些特定的角色進(jìn)行了定義。根據(jù)歐盟MDR和MPD���,企業(yè)須為器械任命至少一位監(jiān)管合規(guī)負(fù)責(zé)人(PRRC)�����,為藥品配備一名符合條件的藥物警戒負(fù)責(zé)人(QPPV)和至少一名質(zhì)量負(fù)責(zé)人(QP)���。監(jiān)管合規(guī)負(fù)責(zé)人、藥物警戒負(fù)責(zé)人和質(zhì)量負(fù)責(zé)人在質(zhì)量管理和產(chǎn)品安全方面發(fā)揮關(guān)鍵作用��,但承擔(dān)的職責(zé)存在較大區(qū)別�����。對(duì)于藥品�����,藥物警戒負(fù)責(zé)人負(fù)責(zé)監(jiān)督和審核藥品的安全性,質(zhì)量負(fù)責(zé)人負(fù)責(zé)保證藥品的安全性��、質(zhì)量(批次控制)和療效��。對(duì)于醫(yī)療器械�����,監(jiān)管合規(guī)負(fù)責(zé)人對(duì)醫(yī)療器械的安全性��、質(zhì)量和監(jiān)管合規(guī)負(fù)責(zé)���,這似乎結(jié)合了藥物警戒負(fù)責(zé)人和質(zhì)量負(fù)責(zé)人的角色��。
表1:根據(jù)歐盟MDR和MPD�����,監(jiān)管合規(guī)負(fù)責(zé)人���、藥物警戒負(fù)責(zé)人和質(zhì)量負(fù)責(zé)人的監(jiān)管職責(zé)概覽
如上表所示��,盡管這三個(gè)角色均負(fù)責(zé)確保產(chǎn)品的安全性��、質(zhì)量和有效性符合相關(guān)法規(guī)���,各角色的職責(zé)范圍又各不相同。因此�����,必須要有清晰的崗位描述和過(guò)程�����,確保相關(guān)職責(zé)得到了充分的履行��。
結(jié)論
制藥企業(yè)在研發(fā)組合產(chǎn)品時(shí)��,實(shí)施醫(yī)療器械QMS體系會(huì)帶來(lái)明顯的優(yōu)勢(shì)。然而�����,這需要仔細(xì)的考量和策略性的規(guī)劃��。針對(duì)新器械/未來(lái)器械的新能力(包含相應(yīng)專(zhuān)業(yè)知識(shí))���、允許專(zhuān)業(yè)人員根據(jù)相關(guān)要求執(zhí)行任務(wù)的新過(guò)程���,以及處理醫(yī)療器械要求的新IT系統(tǒng),都會(huì)是制藥企業(yè)實(shí)施醫(yī)械QMS體系的必需項(xiàng)���。因此�����,利益相關(guān)者的管理在整個(gè)過(guò)程(從規(guī)劃到實(shí)施)都至關(guān)重要���。此外,制藥企業(yè)將需要與相關(guān)區(qū)域的不同監(jiān)管機(jī)構(gòu)以及公告機(jī)構(gòu)(歐盟)建立新的關(guān)系��。
英文原文
The global market for drug-device combination products is booming; it was valued at $118.13 billion in 2021 and is expected to expand at a compound annual growth rate of 8.8% by 2030. To capture the growing market with minimal business disruption, most pharma companies used to rely on third parties to develop and obtain the regulatory approval for the device in a combination product, a product that combines a medicinal product or substance and a medical device in a single integral unit (e.g., prefilled syringes). However, pharma companies are now building the capability to manufacture combination products in-house to increase flexibility and reduce dependency on third parties when ensuring the safety and efficacy of the combination product. Therefore, pharma companies have begun to expand their quality management systems (QMS) to allow them to become the legal manufacturer of the medical device components of their combination products. A QMS is a structured system that includes policies, processes, and procedural documents that a pharmaceutical or medical device manufacturer has in place throughout the product’s life cycle. Implementing a QMS that supports medical devices is an essential step in building the new capabilities and becoming a legal manufacturer of such devices.
This will also enable pharma companies to manufacture personalized treatments that maximize positive health outcomes and system resilience, which is a market that will increase by $70 billion to $390.4 billion by 2030. In this article we discuss the challenges and considerations for companies that are interested in entering the EU market with combination products.
How Are Combination Products Regulated?
Combination products are governed by Regulation (EU) 2017/745 (EU MDR) as a medical device or by the Medicinal Products Directive 2001/83/EC (MPD) as a drug, depending on the principal mode of action.
The EU Medical Device Coordination Group (MDCG) recently published guidance document MDCG 2022-5, Guidance on borderline between medical devices and medicinal products under EU MDR, providing additional information for manufacturers when deciding the regulatory status of a combination product.
When a combination product is considered a drug, and thus regulated under the MPD, the medical device in the combination still needs to meet specific requirements of EU MDR. Examples of combination products regulated as medicinal products are prefilled syringes (e.g., vaccine in a prefilled syringe) and single-use inhalers. Having a QMS that complies with both pharma and device regulatory requirements will ensure that the combination product will meet the requirements of both regulations.
Advantages Of Implementing A Medical Device QMS
The major benefit of having a medical device QMS is that the device development and related documentation will comply with the applicable regulatory requirements and the company can CE mark1 the device to demonstrate compliance of the device with EU MDR requirements. The letters “CE” signify that products sold in the European Economic Area (EEA) have been assessed to meet high safety, health, and environmental production requirements.
When a device is developed and CE marked by a third party, the pharma company has less oversight of the process and timelines. By becoming a legal manufacturer of medical devices, the pharma company can CE mark the devices by issuing a Declaration of Conformity per their own project plan and will be less dependent on third parties.
If the device is not CE marked or the device is high-risk, the company has to submit a dossier to a Notified Body to obtain a Notified Body Opinion that will be part of the Marketing Authorization Application (MAA) for the combination to the European Medicines Agency (EMA). The EMA will expect the submission to comply with EU MDR requirements. Having a medical device QMS in place will ensure the documentation will meet the Notified Body’s expectations.
The advantage of CE marking the device, however, is that it eliminates the need for a Notified Body opinion and takes this approval out of the critical path to submission. The only documentation to be submitted as part of the MAA is the Declaration of Conformity issued by the legal manufacturer, in this case the pharma company.
Combination products are often accompanied by an app to enhance the patient’s or healthcare professional’s experience. Depending on the features of the app, it can also qualify as medical device software (SaMD) and will need CE marking to be marketed in the EU. Having a medical device QMS in place will allow a pharma company to develop and CE mark both the device in the combination and the stand-alone app.
Considerations For Implementing A Medical Device QMS In A Pharmaceutical Environment
Implementing a medical devices QMS in a pharmaceutical environment requires careful consideration and planning to do it right the first time and avoid any delays or additional costs.
Therefore, it is key to perform an end-to-end assessment of the existing processes, capabilities, and systems to identify any gaps, as well as the resources that can be leveraged to build a medical device QMS. Building the medical device QMS as an extension of the pharma QMS has the advantage that existing processes and resources can be leveraged. Obtaining ISO 13485 certification for the QMS from an independent certification body can accelerate and harmonize the MDR auditing process, without showing all the product design and documentation to the NB.
The end-to-end assessment needs to include the in-house capabilities to ensure the company has the right expertise to manage and maintain the QMS. For medical device development and post-market surveillance, the company will require experts with biotechnology, biomedical, engineering, software, and related backgrounds. In other words, a company with combination products in its portfolio will need to ensure the employee portfolio includes all necessary expertise to cover both medicinal products and medical devices, so they can keep the QMS up to date.
How To Design A Strategy To Implement A Medical Device QMS
The pharma company must start with a multifunctional project team representing the various parts of the business, including the necessary medical device experts. In many cases, companies will leverage third parties to perform the assessment and to develop the implementation plan.
When considering the document hierarchy of a pharma QMS, the high-level procedural documents, such as policy, can usually be leveraged across devices and drugs because they typically do not contain the details of the process and roles and responsibilities at the execution level. Taking the CAPA (corrective and preventive action) and the complaint management processes as examples, the policy elements will be similar across drugs and devices, whereas the execution will involve different expertise and systems.
Some processes will require enhancement to ensure they are robust across drugs and devices. For example, the risk management process may require different expertise and risk analysis methodologies to address the mechanism of the product. Risk analysis of a medicinal product will mostly focus on the molecular and chemical interaction in the human body associated with a side effect. Risk analysis of a medical device will focus on the design and its use by both the user and the patient in relation to an incident, as medical devices often involve more complex systems. However, the high-level process is very similar for both, as the risk management team will assess the product, score the potential risks, and design a solution to minimize the impact on a patient.
While high-level procedural documents and some processes can be leveraged across different types of products, some lower-level processes may be easier to manage separately. For example, the complaint management process will be easier to manage if there are separate procedures or work instructions for medicinal products and medical devices. This is related to the type of complaints received. For pharma, most of the complaints are related to adverse events and side effects. For medical devices, most of the complaints received are typically quality complaints that do not necessarily have an impact on a patient at the time of reporting. Therefore, complaint evaluation will require different expertise and different processes. Medical devices will typically require a technical assessment of the device’s design and operation, while a medicinal product complaint assessment will require medical/clinical expertise. Therefore, it is essential to have the right experts to ensure a proper investigation is conducted.
Differences In The Regulatory Experts’ Roles
A QMS for medical devices overlaps with pharma QMS requirements but with some significant differences. We use one of the requirements — the regulatory expert’s role — as an example to highlight the commonalities and differences.
Both medical device and medicinal product QMSs require a quality management representative who is responsible for the effectiveness of the QMS; these specific roles are also defined in the regulations. Under the EU MDR and MPD, companies are required to appoint at least one Person Responsible for Regulatory Compliance (PRRC) for devices and have a Qualified Person for Pharmacovigilance (QPPV) and at least one Quality Person (QP) at their disposal for drugs. The PRRC, QPPV, and the QP play key roles in managing the quality and safety of the products, but their responsibilities are quite different. In pharma, the QPPV has the responsibility to monitor and review the safety profiles of the medicinal products and the QP is responsible for assuring the safety, quality (batch control), and efficacy of the medicinal product. For medical devices, the PRRC is responsible for the safety, quality, and regulatory compliance of the medical devices, which seems to be a combination of the roles of the QP and the QPPV.
Table 1: An overview of the regulatory obligations of the PRRC, QPPV, and QP per EU MDR and MPD.
Although all three roles are responsible to ensure the safety, quality, and efficacy of the product comply with the relevant regulations, the scope of each role is quite different, as listed in the table above. Therefore, it is essential to have clear job descriptions and processes in place to ensure the roles are duly fulfilled.
Conclusion
Implementing a medical device QMS in a pharma company will bring significant advantages when developing combination products. However, it needs careful consideration and strategic planning. It will require implementing new capabilities with the right expertise on the new/future medical devices, new processes that will enable the experts to execute in line with the requirements, and new IT systems to address medical device requirements. Therefore, stakeholder management will become essential throughout the entire process, from planning to implementation. In addition, a pharma company will need to establish new relationships with different regulatory groups in a territory and with a Notified Body (EU).
https://www.meddeviceonline.com/doc/combination-product-qms-requirements-for-pharma-companies-entering-the-eu-market-0001
來(lái)源:MED DEVICE ONLINE
作者:Hilde Viroux��、Maggie Chan和Ruby Powell���,均來(lái)自PA Consulting
翻譯:奧咨達(dá)