摘要
在化學藥品注冊中,批量以及生產(chǎn)規(guī)模放大是藥學審評重點關注的問題��。本文在對“批”定義進行剖析的基礎上�,比較了國內外對工藝開發(fā)批量的要求,總結了質量研究����、穩(wěn)定性研究����、非臨床研究��、臨床研究所用批次的批量要求,并提出了進一步完善我國與批量相關的管理和技術要求的建議�。
無論原料藥還是制劑����,工藝開發(fā)的目標是要建立一個能夠持續(xù)��、穩(wěn)定生產(chǎn)出預期質量產(chǎn)品的商業(yè)生產(chǎn)工藝[1-2]��。工藝開發(fā)一般是從實驗室開始,經(jīng)歷生產(chǎn)規(guī)模的放大才能實現(xiàn)商業(yè)生產(chǎn)��。生產(chǎn)規(guī)模放大并非物料量的簡單倍增�,生產(chǎn)場地、生產(chǎn)設備��、操作參數(shù)、物料來源等諸多方面都有可能隨之變更,并對產(chǎn)品質量產(chǎn)生潛在的影響�。因此,在藥品注冊中,注冊申報的批量以及生產(chǎn)規(guī)模放大是審評機構重點關注的問題之一[1-4]����。近年來,隨著我國化學藥品注冊技術要求的不斷完善����,我國審評機構對藥品注冊中工藝規(guī)模的關注度也在不斷提高[5-7],申報批次的制備批量過小已成為當前發(fā)補中的常見問題��,或是不批準的主要原因之一�。為了提高注冊效率和成功率��,申請人需要準確理解和把握審評機構對于注冊批量的要求��,關注工藝研發(fā)的規(guī)模和工藝放大問題。我們梳理比較了國內外監(jiān)管機構對化學藥品注冊工藝規(guī)模的要求�,分析探討了藥品研發(fā)中主要研究內容所用樣品的批量要求����,供業(yè)界參考����。
一、化學藥品注冊中的“批”定義
ICH 指導原則將“批( batch 或者 lot) ”定義為“在一個或一系列工藝過程中產(chǎn)生的一定量的��、在特定限度內具有均一性的物料”[8]�,這里的“一定量”就是“批量( batch size) ”。通常,原料藥的批量用其實際的批產(chǎn)量來表示,“100 kg 批量”是指每批原料藥的產(chǎn)量在 100 kg 左右; 制劑的批量可以用總物料量或者單位制劑的理論得量來表示����,例如小容量注射液的批量可以表示為“500 L”( 灌裝前的總配液量) 或“10 萬支”(灌裝量為 5 mL����,500 L 的理論灌裝量為 10 萬支) 。
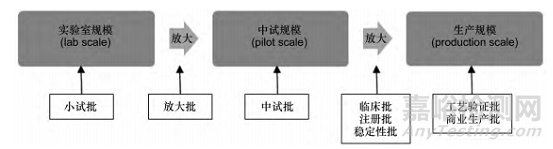
在藥品研發(fā)和注冊中�,根據(jù)“批”的不同用途和相對批量,又衍生出小試批����、中試批、臨床批�、注冊批、商業(yè)批等概念�。以我國新頒布的《化學藥品新注冊分類申報資料要求( 試行) 》為例,其中就涉及注冊批、商業(yè)生產(chǎn)批�、臨床研究批、中試放大批�、生產(chǎn)現(xiàn)場檢查批��、工藝驗證批等概念[7]����。盡管各監(jiān)管機構對衍生出的各種“批”概念所用名詞不盡相同,但在定義上均是根據(jù)批量和用途的不同予以區(qū)分��。見圖 1�。
▲ 圖1-藥品研發(fā)的工藝規(guī)模與批次
1.1 根據(jù)批量相對大小衍生出的“批”概念
基于生產(chǎn)條件和批量的相對大小��,工藝規(guī)模大體上可以分為實驗室規(guī)模�、中試規(guī)模和生產(chǎn)規(guī)模 3 個等級��,相對應的“批”分別為小試批����、中試批和生產(chǎn)批[9]�。小試批(lab scale batch) 是在實驗室條件下生產(chǎn)的批次,受實驗室設備的限制����,批量較小。中試批( pilot scale batch) 是采用中試規(guī)模和設備生產(chǎn)的批次,ICH 指導原則將其定義為“采用能夠充分代表和模擬將用于最大生產(chǎn)規(guī)模的工藝所生產(chǎn)的批次; 對于固體口服制劑��,中試規(guī)模通常至少為最大生產(chǎn)規(guī)模的 1 /10 或者 10 萬制劑單位( 片或膠囊) 中的較大者”[10]。生產(chǎn)批( production batch) 對應于生產(chǎn)規(guī)模����,是在注冊申請中界定的生產(chǎn)設施中使用大生產(chǎn)設備生產(chǎn)的批次[10]。
需要注意的是����,工藝規(guī)模只是針對具體產(chǎn)品反映批量相對大小的一個概念。對一個產(chǎn)品而言����,各規(guī)模對應的批量不是唯一的��,例如一個片劑����,其生產(chǎn)規(guī)?���?赡苡?25����,50����,100 萬片等多種批量; 對于不同產(chǎn)品而言,各規(guī)模對應的批量也不具可比性����,例如同為生產(chǎn)規(guī)模��,一些原料藥的批量可能達到幾百公斤����,而也有一些原料藥可能僅有幾十克。
另外�,在藥品研發(fā)過程中��,根據(jù)產(chǎn)品和工藝特點�,在小試批和中試批之間��、中試批和大生產(chǎn)批之間還可能有中間狀態(tài)的放大批( scale-up batch) ; 在一些情況下,例如商業(yè)生產(chǎn)的批量較小����,或者產(chǎn)品的生產(chǎn)工藝非常簡單,也有可能不需要經(jīng)過中試放大��,直接從實驗室規(guī)模過渡到生產(chǎn)規(guī)模����。
1.2 根據(jù)用途不同衍生出的“批”概念
根據(jù)“批”的用途不同又衍生出臨床批、工藝驗證批��、商業(yè)批等概念,監(jiān)管機構通常會對不同用途的批次提出各自的批量要求。例如�,臨床批 ( clinical batch����,或 者biobatch) 是指用于臨床研究、人體生物利用度或生物等效性等研究的批次�,至少需要中試以上規(guī)模[11]����。在傳統(tǒng)工藝驗證理念下�,工藝驗證批( process validation batch) 是指在商業(yè)生產(chǎn)前��,為驗證工藝,按照確定的生產(chǎn)工藝在商業(yè)生產(chǎn)規(guī)模上所生產(chǎn)的批次�。商業(yè)批( commercial batch) 是指藥品獲準上市后��,采用生產(chǎn)規(guī)模生產(chǎn)并用于商業(yè)銷售的批次��。
國際上將用于正穩(wěn)定性研究的批次稱為“primary batch”或者“primary stability batch”,從這些批次中產(chǎn)生的穩(wěn)定性數(shù)據(jù)用于支持建立原料藥的再檢驗期或制劑的有效期[10]; 在我國頒布的《化學藥物( 原料藥和制劑) 穩(wěn)定性研究技術指導原則》中����,則將這些用于正式穩(wěn)定性研究的批次稱為“注冊批”[12]。另外�,在我國頒布的《化學藥品新注冊分類申報資料要求( 試行) 》等中也提出了“注冊批”概念[6-7]����,但未給出“注冊批”的明確定義�,從指導原則內容來看����,此處的“注冊批”與上述穩(wěn)定性研究技術指導原則中的“注冊批”內涵不完全相同��,而和 FDA 指導原則中所指的“展示批( exhibit batch) ”[13-15]含義較為接近,這些批次的工藝信息�、生產(chǎn)數(shù)據(jù)����、中間體和成品檢驗結果等都是支持注冊申請的重要信息; 這些批次如果符合穩(wěn)定性研究對批次的要求并用于正式的穩(wěn)定性研究����,方能稱之為“primary batch”。
在研發(fā)過程中����,同一批次可以有多種用途��。例如��,只要能夠符合法規(guī)、指導原則對相關研究所用批次的規(guī)定和要求�,“注冊批”可以用于穩(wěn)定性�、生物等效性或臨床等研究��。需要指出的是��,我國一些申請人經(jīng)常在中試規(guī)模生產(chǎn)“注冊批”時同步開展工藝驗證,并把“注冊批”也稱為“工藝驗證批”��。盡管采用中試規(guī)模開展的工藝驗證研究可以視為工藝驗證生命周期的組成部分�,但傳統(tǒng)意義上的工藝驗證通常是在產(chǎn)品上市之前采用生產(chǎn)規(guī)模開展����,此時產(chǎn)品和工藝開發(fā)已經(jīng)完成�,工藝也已經(jīng)放大到生產(chǎn)規(guī)模[16]�。由于中試規(guī)模的“注冊批”并不符合傳統(tǒng)工藝驗證對批次的要求��,稱之為“工藝驗證批”不夠嚴謹��,申請人仍需要按照指導原則要求采用前三批商業(yè)生產(chǎn)批開展工藝驗證[7]��。
二����、國內外對化學藥品注冊工藝規(guī)模的要求
盡管工藝放大對產(chǎn)品質量可能產(chǎn)生影響��,但這并不意味著,藥品注冊申報時一定要提供商業(yè)生產(chǎn)規(guī)模的研究資料����。考慮到工藝放大的時間成本等問題��,為縮短產(chǎn)品上市時間�,監(jiān)管機構往往要求在提交新藥申請( NDA) 或者仿制藥申請( ANDA) 時提供的工藝研究信息達到中試規(guī)模即可�。
2.1 國際上對化學藥品注冊工藝規(guī)模的要求
ICH 相關指導原則[1-3]中沒有要求在提交創(chuàng)新藥注冊申請時提供生產(chǎn)規(guī)模的研究數(shù)據(jù)����,但要求工藝研究的規(guī)模應達到中試或以上�。世界衛(wèi)生組織( WHO) 在其仿制藥相關指導原則中也明確說明��,如果提供的工藝信息能夠代表生產(chǎn)規(guī)模,同時承諾在工藝規(guī)模放大時按照變更指導原則的要求及時向 WHO報告��,提供中試規(guī)模的工藝信息是可以接受的[17]��。
美國 FDA 的相關技術文件[14-15,18-20]更為清晰地說明了遞交仿制藥注冊申請時對工藝信息的要求。一般而言�,在仿制藥注冊申請時需要提交中試或以上規(guī)模生產(chǎn)的展示批( exhibit batch) 信息�,明確擬定的商業(yè)生產(chǎn)批量����,說明商業(yè)生產(chǎn)規(guī)模與展示批在批量上的差異�,以及二者所用設備在設計和操作原理方面的異同����,并提交工藝放大計劃�。為了幫助審評員判斷所提交的工藝能否從展示批放大到生產(chǎn)規(guī)模并保持產(chǎn)品質量的一致性�,以及在后續(xù)商業(yè)生產(chǎn)中工藝和產(chǎn)品質量的一致性��,F(xiàn)DA 要求申請人提交詳細的工藝開發(fā)報告以證明申請人對生產(chǎn)工藝有充分的理解。FDA 希望在注冊申報資料中能夠固定生產(chǎn)過程檢驗����、終產(chǎn)品放行標準以及工藝描述��,但是允許申請人在從展示批向生產(chǎn)規(guī)模的放大過程中對操作參數(shù)( 例如時間��、流速����、溫度等) 進行調整,以滿足上述已固定的各項要求。FDA 要求申請人在申報資料中說明擬采用的商業(yè)生產(chǎn)規(guī)模的操作參數(shù)��,并與關鍵批次( 例如用于生物等效性研究的展示批) 的相應參數(shù)進行比較��、分析�。對于擬采用的生產(chǎn)規(guī)模的操作參數(shù),申請人在申報時既可以將其固定下來,也可以說明目前僅是臨時參數(shù)����,將根據(jù)后續(xù)放大研究進行調整。見表 1�。
▲ 表1-FDA 對不同規(guī)模工藝參數(shù)的比較要求
總體而言��,以 FDA、EMA 為代表的國外監(jiān)管機構并不強制要求在注冊申請時提交生產(chǎn)規(guī)模的研究數(shù)據(jù)�,但非常關注申報批次的批量與商業(yè)生產(chǎn)規(guī)模的批量差異����,以及工藝放大的風險評估和風險管理[9,16����,18-21]��。為了有效控制用較小規(guī)模(例如中試規(guī)模) 研究數(shù)據(jù)支持產(chǎn)品注冊的潛在風險,這些監(jiān)管機構除強調注冊申請資料中相關工藝研究信息的提交要求外����,更重要的是針對上市后生產(chǎn)規(guī)模放大建立了較為完善的監(jiān)管措施[22-25]�。
2.2 我國對化學藥品注冊工藝規(guī)模的要求
我國于 2005 年頒布的相關技術指導原則中闡述了化學原料藥和制劑的工藝放大研究要求��,但沒有明確注冊申報時對工藝規(guī)模的要求����,也沒有明晰“中試規(guī)模”的定義[26-27]�。由于指導原則對注冊申報的工藝規(guī)模缺少明確要求,加之當時我國藥品研發(fā)機構對工藝研究包括規(guī)模問題的重要性認識不足�,一些申報品種的研制規(guī)模很小�,對工藝的認知不夠深入,導致申報工藝不能順利過渡到商業(yè)生產(chǎn)��,在產(chǎn)品獲批后,需要另起爐灶重新開發(fā)適合商業(yè)生產(chǎn)的工藝����,加之后續(xù)的變更管理沒有跟上�,造成申報工藝與實際生產(chǎn)工藝不一致的問題�。
隨著我國藥品研發(fā)水平的提升,對工藝規(guī)模的重要性有了新的認知��。2008 年頒布的《化學藥品技術標準》提出了“規(guī)?�;a(chǎn)的可行性”問題,強調“原料藥的制備工藝研究應在一定規(guī)模下開展����,所取得的研究數(shù)據(jù)( 包括工藝條件�、工藝參數(shù)�、起始原料和中間體的質量控制要求等) 應能直接用于或指導原料藥的工業(yè)化生產(chǎn)”����,對于制劑也要求“臨床試驗( 含生物等效性試驗) 用樣品的處方工藝應與實際生產(chǎn)產(chǎn)品的處方工藝一致����,現(xiàn)制備規(guī)模下的產(chǎn)品質量應能代表工業(yè)化生產(chǎn)的產(chǎn)品質量”����,對于工藝規(guī)模過小的原料藥和制劑申請不予批準[5]。該要求的頒布使研發(fā)人員和審評人員逐漸開始重視工藝規(guī)模問題����。
2010 年我國開始對部分化學藥品注冊申請試行 CTD 格式申報����,引入了“注冊批”�、“大生產(chǎn)的擬定批量范圍”等概念[6]��,技術審評中對批量問題的重視程度進一步提高,開始關注生產(chǎn)工藝開發(fā)過程中的規(guī)模變化及其對產(chǎn)品質量的影響��、注冊申報規(guī)模與擬定商業(yè)生產(chǎn)規(guī)模的差異及其潛在風險等����,這與國際上對工藝規(guī)模的要求逐漸趨于一致����。最近頒布的《化學藥品新注冊分類申報資料要求( 試行) 》��,對于新注冊分類 1����、2��、3 和 5.1 仍延續(xù)了上述表述和要求����,允許注冊批規(guī)模與擬定的大生產(chǎn)規(guī)模存在一定差距; 對于新注冊分類 4 及 5.2,則進一步提高了要求�,明確提出“擬定的大生產(chǎn)的批量范圍不能超出研發(fā)過程中的最大生產(chǎn)批量”����,否則需要提供充分的放大研究與驗證的依據(jù)。另外����,該申報資料要求中也對“中試規(guī)模”進行了定義�,無論原料藥和制劑,要求中試規(guī)模的批量至少為商業(yè)化生產(chǎn)規(guī)模的1 /10[7]�,這與國際上通用的定義有所不同��。
三��、一些研究用樣品的批量要求
為了保證商業(yè)生產(chǎn)產(chǎn)品與研發(fā)過程中關鍵批次樣品的質量、安全性�、有效性保持一致����,監(jiān)管機構對藥品研發(fā)中用于關鍵的藥學、非臨床和臨床研究的樣品批量往往也會提出要求����。
3.1 質量研究
“質量研究”是我國藥品注冊中沿用多年的一個概念��,其內容包括確定研究項目��、建立檢測方法并進行方法學驗證、制訂質量標準并建立各檢測項目的限度要求等�。我國指導原則要求“藥物質量研究一般需采用試制的多批樣品進行,其工藝和質量應穩(wěn)定”�,臨床前的質量研究工作要求采用“一定規(guī)模制備的樣品( 至少 3 批) ”進行,臨床研究期間對“中試或工業(yè)化生產(chǎn)規(guī)模的多批樣品”進行質量研究工作[28]�。ICH 以及 FDA����,EMA 等發(fā)布的技術文件中沒有出現(xiàn)“質量研究”的概念��,與我國“質量研究”相對應的研究內容包括雜質��、殘留溶劑����、重金屬研究以及分析方法驗證����、質量標準制定等�,與之相關的指導原則中也沒有特別明確對研究用樣品規(guī)模的要求��。
對于分析方法驗證����,其部分內容例如專屬性、準確度可能和所用樣品的生產(chǎn)規(guī)模相關�,這是由于生產(chǎn)規(guī)模的變化可能導致雜質譜的變化����,或者引起制劑處方等方面的關聯(lián)變更��,進而影響方法的專屬性、準確度����,而其他一些驗證內容例如線性����、范圍��、定量限等則與樣品的生產(chǎn)規(guī)模不相關。因此�,如果采用較小規(guī)模生產(chǎn)的樣品進行分析方法開發(fā)及方法驗證,當生產(chǎn)規(guī)模放大時����,需要評估規(guī)模變化及其他關聯(lián)變更對分析方法的影響,必要時對分析方法的專屬性�、準確度等進行確認或再驗證��。如果規(guī)模的變化導致產(chǎn)品中出現(xiàn)了新的需要單獨控制的雜質�,則需要針對這些雜質進行方法的全驗證。
不同規(guī)模�、不同用途樣品的批分析數(shù)據(jù)是建立質量標準限度的重要依據(jù)之一����。對于早期研發(fā)階段的創(chuàng)新藥����,由于生產(chǎn)規(guī)模較小��,更多是依據(jù)小規(guī)模生產(chǎn)的非臨床/臨床批次樣品的檢測數(shù)據(jù)��,結合相關的開發(fā)數(shù)據(jù)、藥典和指導原則要求等來制訂限度; 在NDA 和 ANDA 申請中�,檢測項目及限度的設定則要充分考慮放大規(guī)模生產(chǎn)樣品的檢測結果和穩(wěn)定性數(shù)據(jù)[29]�。
3.2 穩(wěn)定性研究
對于藥品注冊申報時正式穩(wěn)定性研究所用樣品的生產(chǎn)規(guī)模要求,我國與 ICH 基本一致����,其中原料藥應包括至少 3 批中試或以上規(guī)模生產(chǎn)的樣品,新制劑的 2 批樣品應在中試或以上規(guī)模����,第 3 批規(guī)?���?梢孕∫恍?0,12]�。對于仿制制劑( 新注冊分類 4 及 5.2) ,我國的最新要求是穩(wěn)定性批次均應達到中試或以上規(guī)模[7]����,而國際上則多沿用新制劑的要求��。例如����,F(xiàn)DA 要求仿制藥中的口服制劑�,穩(wěn)定性研究批次中至少 2 個批次達到中試或以上規(guī)模,第 3 個較小批次可以小于擬定大生產(chǎn)規(guī)模的 10% ��,但不能低于中試規(guī)模的 25%[30]��。WHO的要求相對更低����,仿制制劑的穩(wěn)定性研究僅需采用每個規(guī)格不少于 2 批至少中試規(guī)模樣品即可; 對于非復雜制 劑( 例如速釋固體制劑、非滅菌溶液劑等) �,其中一批的規(guī)模可以更小( 例如��,對于固體口服制劑 25 000 或 50 000 片/粒) [17]����。
需要指出的是,我國頒布的相關指導原則[7����,12]中對于中試規(guī)模( pilot scale) 的定義和要求與 ICH指導原則存在差異��。ICH 指導原則中僅針對固體口服制劑舉例�,明確中試規(guī)模一般是指最大生產(chǎn)規(guī)模的 1 /10 或 100 000 個制劑單位中的較大者����,而我國的指導原則中對原料藥和制劑均明確“批量至少為商業(yè)化生產(chǎn)規(guī)模的 1 /10”�,同時去除了固體口服制劑 10 萬個制劑單位的要求��。這種變化顯然是考慮到中國醫(yī)藥產(chǎn)業(yè)市場集中度不高的現(xiàn)狀,從可操作性角度而言具有一定合理性��。
3.3 非臨床研究
創(chuàng)新藥研發(fā)是一個漸進的過程,大部分的非臨床研究在研發(fā)早期開展,此時藥學研究包括工藝研究尚不成熟����,生產(chǎn)規(guī)模也比較小��,藥品注冊中對于創(chuàng)新藥非臨床研究用樣品的制備規(guī)模沒有明確限定��。一般而言����,在這個階段�,樣品的生產(chǎn)規(guī)模能夠滿足相應的非臨床和臨床研究需求即可����,根據(jù)具體品種的情況,可以在實驗室規(guī)?�;蚍糯笠?guī)模下生產(chǎn)研究用樣品����。
在創(chuàng)新藥研發(fā)早期階段,臨床研究用樣品的質量標準很大程度上要依據(jù)非臨床研究特別是安全性評價用樣品的檢測結果設定����,對于雜質而言,超過鑒定限的雜質限度原則上不能超出非臨床安全性評價樣品所含相應雜質的實測結果��。在從非臨床向臨床過渡的過程中�,如果生產(chǎn)規(guī)模變化過大,雜質譜發(fā)生變化的可能性會增大��,在確定非臨床研究樣品生產(chǎn)規(guī)模時需要考慮這種風險�。
3.4 臨床研究
藥品注冊中�,用于支持藥品上市的安全性、有效性數(shù)據(jù)并不是來源于商業(yè)生產(chǎn)批次��,而是來源于研發(fā)過程中的有限批次��,例如用于創(chuàng)新藥關鍵的Ⅲ期臨床試驗或者用于仿制藥人體生物等效性試驗的批次。藥品需要在其生命周期內保持與關鍵性臨床研究所用樣品質量的一致性����,這是保證商業(yè)生產(chǎn)批次產(chǎn)品與臨床研究用樣品具有相同安全性����、有效性的基礎�,建立關鍵臨床研究批次與商業(yè)生產(chǎn)批次之間的質量聯(lián)接��,使二者質量保持一致是藥學審評的一個重點關注問題[31]��。但是�,從臨床批次過渡到商業(yè)批次可能有諸多方面的變更�,包括生產(chǎn)規(guī)模的變更�,這些變更會對藥品質量產(chǎn)生潛在的影響。為了降低或避免這種風險��,各監(jiān)管機構非常關注商業(yè)生產(chǎn)批次與關鍵的臨床批次之間在生產(chǎn)工藝包括批量等方面的差異��,申請人需要對這種差異進行充分的討論�,分析差異對產(chǎn)品的生產(chǎn)�、性能和質量的影響[1-3]。
對于創(chuàng)新藥早期研發(fā)階段,臨床研究用樣品可能在小規(guī)模下生產(chǎn)����,監(jiān)管機構會要求申請人提供相關生產(chǎn)信息�,但通常不會關心規(guī)模的大小�。當創(chuàng)新藥進入關鍵的臨床研究( 例如Ⅲ期臨床研究) �,監(jiān)管機構開始關注生產(chǎn)規(guī)模,此時臨床研究用樣品的生產(chǎn)規(guī)模一般應達到中試或以上規(guī)模�,以保證與將來商業(yè)生產(chǎn)的有效橋接[32-33]�。對于仿制藥的口服制劑����,除非有特別理由����,通常要求用于生物等效性研究的樣品批量也應達到中試或以上規(guī)模,即至少為最大生產(chǎn)規(guī)模的 1 /10 或者 10 萬制劑單位中的較大者[34-35]。
四��、討論與建議
藥品注冊的批量問題是當前階段困擾我國藥品研發(fā)和審評的一個重要問題��,這一問題的產(chǎn)生和我國醫(yī)藥產(chǎn)業(yè)的發(fā)展現(xiàn)狀有很大關系��。從研發(fā)角度而言����,由于我國醫(yī)藥生產(chǎn)企業(yè)普遍規(guī)模較小�,同品種重復申報嚴重,加上流行病學數(shù)據(jù)的缺失、諸多政策性因素的影響��,申請人在進行研發(fā)時很難預測將來的市場占有量��,也就很難確定商業(yè)生產(chǎn)規(guī)模及工藝的研發(fā)規(guī)模; 另外��,研發(fā)成本和工藝開發(fā)的規(guī)模有很大的相關性�,由于研發(fā)期間制備的產(chǎn)品不能上市銷售��,為了降低研發(fā)成本����,申請人不斷試探審評機構對于工藝規(guī)模要求的底線�,盡可能降低工藝研發(fā)的規(guī)模。從審評角度而言�,審評機構需要根據(jù)提交的工藝研究、生產(chǎn)及質量控制信息��,判斷申請人是否能夠從現(xiàn)有規(guī)模放大到商業(yè)生產(chǎn)規(guī)模并保持產(chǎn)品質量的一致性�,以及是否能夠在商業(yè)生產(chǎn)中持續(xù)穩(wěn)定地生產(chǎn)出質量一致的產(chǎn)品; 工藝研究越充分,工藝規(guī)模越接近商業(yè)生產(chǎn)規(guī)模����,審評機構做出判斷的難度越小,風險也越小��,因此審評機構希望申請人在注冊申報時提供與生產(chǎn)規(guī)模較為接近的工藝研發(fā)數(shù)據(jù)。
批量問題實質上是生產(chǎn)規(guī)模變更的“風險”問題��,完善由研發(fā)到生產(chǎn)工藝規(guī)模變更的風險控制��,有助于申請人與審評機構在批量問題上達成共識��?;谇拔牡姆治觯覀兘ㄗh: ① 進一步明晰我國相關法規(guī)文件及指導原則中所列出的各種“批”的定義和內涵�,包括對規(guī)模的要求。② 借鑒其他監(jiān)管機構做法��,要求申請人基于研發(fā)期間的數(shù)據(jù)分析工藝的規(guī)模放大效應����,并結合類似工藝的經(jīng)驗分析工藝放大的風險,比較研發(fā)規(guī)模與擬定的生產(chǎn)規(guī)模在物料�、設備、參數(shù)等方面的差異����,提出批量放大計劃; 審評中應重點關注申報批次和擬定商業(yè)生產(chǎn)規(guī)模的批量差異及潛在風險,而非聚焦于批量的“絕對大小”����。③ 生產(chǎn)規(guī)模變更是常見的上市后變更情形之一����,但在我國現(xiàn)行法規(guī)�、指導原則中對上市后生產(chǎn)規(guī)模變更的管理要求和技術要求較為模糊,現(xiàn)實中對于上市后的生產(chǎn)規(guī)模變更也缺乏有效監(jiān)管��,可考慮在藥品注冊批準文件中注明現(xiàn)有研發(fā)數(shù)據(jù)可支持的生產(chǎn)規(guī)模��,同時加強上市后規(guī)模變更的管理�,根據(jù)規(guī)模變更對產(chǎn)品質量的影響程度設置不同的管理通道,例如年度報告�、備案、補充申請等�。
參考文獻
[1]ICH.Harmonised Tripartite Guideline Q8(R2):Pharmaceutical development [EB /OL].[2009-08-01]( 2016-05 -11).http: / /www.ich.org/fileadmin /Public _Web_Site/ICH_Products/Guidelines/Quality/Q8_1/Step4/Q8_R2_Guideline.pdf.
[2]ICH.Harmonised Tripartite Guideline Q11 : Development and manufacture of drug substances ( chemical entities and biotechno logical / biological entities) [EB /OL].[2012-05-01]( 2016-05-11) . http: / /www.ich.org /fileadmin /Public_Web_Site / ICH_Products/Guidelines/Quality /Q11 /Q11_Step_4.pdf.
[3]ICH. Harmonised Tripartite Guideline M4Q( R1) : The common technical document for the registration of pharmaceuticals humanuse: quality [EB /OL].[2002-09-12]( 2016-05-11) .http: / /www.ich.org /fileadmin /Public _ Web _ Site / ICH _ Products/CTD /M4_R1_Quality /M4Q__R1_. pdf.
[4]FDA. Manual of policies and procedures: Chemistry Review of Question-based Review ( QbR) Submissions[EB /OL].[2014-11-18]( 2016-04-17) .http://www.fda.gov/downloads/aboutfda/centersoffices/officeofmedicalproductsandtobacco /cder /manualofpoliciesprocedures/ ucm423752.pdf.
[5] 國家食品藥品監(jiān)督管理總局.化學藥品技術標準[EB /OL].[2008 -06-03]( 2016-04-21). http: / /www.sda.gov.cn /WS01 /CL0844 /30470.html.
[6] 國家食品藥品監(jiān)督管理總局.化學藥品 CTD 格式申報資料撰寫要求 [EB /OL].[2010- 09 -25]( 2016 -04-21) .http: / /www.sda.gov.cn /WS01 /CL0844 /54391. html.
[7] 國家食品藥品監(jiān)督管理總局.化學藥品新注冊分類申報資料要求( 試行) [EB /OL].[2016-05- 04]( 2016-05-04).http: / /www.sda.gov.cn /WS01 /CL0087 /151985.html.
[8] ICH.Harmonised Tripartite Guideline Q7: Good manufacturing practice guide for active pharmaceutical ingredients [EB /OL]-[2000-10-10]( 2016-04-15 ) .http: / /www.ich.org /fileadmin /Public_Web _ Site / ICH _ Products/Guidelines/Quality /Q7 / Step4 /Q7_Guideline- pdf-
[9] EMA.Note for guidance on process validation[EB/OL].[2001-03-01] ( 2016-04-15 ) -http: / /www.ema.europa.eu /docs/en _ GB / document _ library / Scientific _ guideline /2009 /09 /WC500002913.pdf.
[10] ICH.Harmonised Tripartite Guideline Q1A( R2) : Stability testing of new drug substances and products [EB /OL].[2003-02-06]( 2016-04-15 ) .http: //www.ich.org/fileadmin/Public_Web _ Site / ICH _Products/Guidelines/Quality /Q1A _R2 /Step4 /Q1A_R2__Guideline.pdf.
[11] EMA.Guideline on quality of oral modified release products[EB /OL].[2014-03-20](2016-04-21) . http: / /119.90.25.46 /www.ema.europa.eu / docs/en_GB / document_library / Scientific_guideline /2014 /07 /WC500170465.pdf.
[12] 國家食品藥品監(jiān)督管理總局.化學藥物( 原料藥和制劑) 穩(wěn)定性研究技術指導原則 [EB /OL]. [2015-02-05]( 2016-04-21).http: / /www.sda. gov. cn/WS01/CL1616/114289. html.
[13] FDA.Guidance for Industry: Powder Blends and Finished Dosage Units — Stratified In-Process Dosage Unit Sampling and Assessment ( Draft guidance) [EB/OL].[2003-10-01](2016-04-21) .http: / /www.fda.gov/ohrms/ dockets/98fr/03d-0493-gdl0001.pdf.
[14] FDA.Quality by Design for ANDAs: An Example for ModifiedRelease Dosage Forms [EB /OL].[2011-12-01]( 2016-04-21) .
http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM286595.pdf.
[15] FDA. Quality by Design for ANDAs: An Example for Immediate-Release Dosage Forms [EB /OL].[2012-04-01]( 2016-04-21 ). http://www.fda.gov/downloads/Drugs/Development ApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM304305.pdf.
[16] EMA.Guideline on process validation for finished products-information and data to be provided in regulatory submissions [EB /OL].[2014-02-27]( 2016-04-21). http: / /www. ema.europa.eu / docs/en_GB / document_library / Scientific_guideline /2014 /02 /WC500162136. pdf.
[17] WHO. Guidelines on submission of documentation for a multisource ( generic) finished pharmaceutical product for the WHO Prequalification of Medicines Programme: quality part [EB /OL].WHO Technical Report Series No. 970,2102. ( 2016-04-21 ).http: / /apps.who.int / prequal /info _ general / documents/TRS970 /TRS_970-Annex4.pdf.
[18] FDA.QbR Frequently asked questions[EB/OL].[2007-06-04]( 2016-04-29 ) .
http://www.fda.gov/downloads/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved /approvalapplications/abbreviatednewdrugapplicationandagenerics/ucm120980.pdf.
[19] FDA.Example quality overall summary [EB/OL].[2009-03-16]( 2016-05-21) .
http://www.fda.gov/downloads/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved /approvalapplications/abbreviatednewdrugapplicationandagenerics/ucm120979. pdf.
[20] FDA.Example quality overall summary [EB/OL].[2009-03-16]( 2016-05- 21).
http://www.fda.gov/downloads/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/abbreviatednewdrugapplicationandagenerics/ucm120977.pdf.
[21] ICH.ICH Quality implementation working group points to consider (R2) : ICH-Endorsed Guide for ICH Q8 /Q9 /Q10 Implementation [EB /OL]. [2011-12-06]( 2016-04-1) .http: / /www.ich.org /fileadmin /Public_Web_Site / ICH_Products/Guidelines/Quality /Q8 _ 9 _ 10 _ QAs/PtC /Quality _ IWG _ PtCR2 _6dec2011. pdf.
[22] FDA. Changes to an Approved NDA or ANDA: Questions andAnswers[EB /OL]. [2001-01-01]( 2016-04-21) . http: / /www. fda. gov / downloads/Drugs/GuidanceComplianceRegulatoryInformation /Guidances/UCM122871. pdf.
[23] FDA. SUPAC-IR: Immediate Release Solid Oral Dosage Forms:Scale-up and Postapproval Changes: Chemistry����,Manufacturing�,and Controls,In Vitro Dissolution Testing����,and In Vivo Bioequivalence Documentation [EB /OL].[1995-11-01]( 2016-04-21 ) .
http://wwwfdagovdownloads/Drugs/Guidance ComplianceegulatoryInformation/Guidances/UCM070636. pdf.
[24] FDA.SUPAC-MR: Modified Release Solid Oral Dosage Forms Scale-up and Postapproval Changes: Chemistry,Manufacturing����,and Controls; In Vitro Dissolution Testing and In Vivo Bioequivalence Documentation [EB /OL].[1997-09-01]( 2016-04-21 ).http: / /www.fda.gov / downloads/Drugs/Guidance ComplianceRegulatoryInformation /Guidances/UCM070640. pdf.
[25] EMA.. Guideline on the details of the various categories of variations to the terms of marketing authorizations for medicinal products for human use and veterinary medicinal products[EB /OL].[2013-08-02]( 2016-04- 21 ) .
http: / /ec. europa. eu /health/files/eudralex /vol-2 /c_2013_2008 /c_2013_2008_pdf /c_2013_2804_en. pdf.
[26] 國家食品藥品監(jiān)督管理總局. 化學藥物原料藥制備和結構確證研究的技術指導原則[EB /OL]. [2005-03 -18]( 2016-04-25) . http: / /www. sda. gov. cn /gsz05106 /02. pdf.
[27] 國家食品藥品監(jiān)督管理總局. 化學藥物制劑研究基本技術指導原則[EB /OL]. [2005 - 03 - 18]( 2016-04-25 ) . http: / /www. sda. gov. cn /gsz05106 /04. pdf.
[28] 國家食品藥品監(jiān)督管理總局. 化學藥物質量標準建立的規(guī)范化過程技術指導原則[EB /OL]. [2005- 03-18]( 2016-04-25) . http: / /www. sda. gov. cn /gsz05106 /16. pdf.
[29] ICH. Harmonised Tripartite Guideline Q6A: Specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances [EB /OL].[1999 -10-06]( 2016-04-15 ).
http: / /www.ich.org /fileadmin /Public _ Web _ Site / ICH _ Products/Guidelines/Quality /Q6A /Step4 /Q6Astep4.pdf.
[30] FDA.ANDAs: Stability Testing of Drug Substances and Products-Questions and Answers[EB /OL].[2014-05-01]( 2016-05-15 ).
http: / /www.fda.gov / downloads/ drugs/guidancecomplianceregulatoryinformation /guidances/ ucm366082.pdf.
[31] 陳震. 我國化學藥品注冊藥學研究技術要求的發(fā)展[J]. 中國新藥雜志�,2014��,23( 1) : 20 - 24.
[32] FDA.Guideline on the preparation of investigational new drug products ( human and animal) [EB /OL].[1991-03-01]( 2016-05-15 ) .http: / /www. fda. gov / downloads/Drugs/GuidanceComplianceRegulatoryInformation /Guidances/UCM070315. pdf.
[33]FDA.INDs for Phase 2 and Phase 3 Studies: Chemistry��,Manufacturing����,and Controls Information [EB / OL].[2003-05-01] ( 2016-05-15 ).http: / /www.fda.gov / downloads/Drugs/GuidanceComplianceRegulatoryInformation /Guidances/UCM070567.pdf.
[34] FDA.Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs — General Considerations ( draft guidance) [EB / OL].[2014-03-01](2016-05 -15) .http: / /www.fda.gov / downloads/Drugs/GuidanceComplianceRegulatory Information /Guidances/UCM389370.pdf.
[35]EMA.Guideline on the investigation of bioequivalence[EB /OL].[2010-01-20]( 2016-05-15).http: / /www.ema.europa.eu / docs/en _ GB / document _ library / Scientific _ guideline /2010 /01 /WC500070039.pdf.