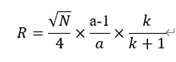藥品是特殊商品,安全、有效�����、質(zhì)量可控是其內(nèi)在屬性。影響藥品安全性的關(guān)鍵是雜質(zhì)�����,包括無(wú)機(jī)雜質(zhì)�����、有機(jī)雜質(zhì)和殘留溶劑�����。建立專屬性強(qiáng)、準(zhǔn)確性高�����、耐用性好的分析方法(圖1)是雜質(zhì)研究與控制的重要手段。其中�����,無(wú)機(jī)雜質(zhì)和殘留溶劑具有通用性,雜質(zhì)種類是明確的�����,因此分析方法的開發(fā)相對(duì)簡(jiǎn)單�����。而有機(jī)雜質(zhì)也就是常說(shuō)的有關(guān)物質(zhì),因API結(jié)構(gòu)和工藝特征而異�����,且多數(shù)有關(guān)物質(zhì)是未知的�����,因此有關(guān)物質(zhì)方法的方法開發(fā)難度更大,是質(zhì)量研究的重頭工作。本文結(jié)合工作經(jīng)驗(yàn)探討有關(guān)物質(zhì)分析方法開發(fā)的流程與技巧�����。
圖1 分析方法目標(biāo)概況(ATP)
在眾多分析方法中�����,高效液相色譜法因具有適用范圍廣�����、操作簡(jiǎn)便的優(yōu)點(diǎn),已成為有關(guān)物質(zhì)測(cè)定最常用的方法�����,因此本文以高效液相色譜法方法開發(fā)為例�����,其他方法也可以類比。
Step1 分析對(duì)象確定
在接到一個(gè)有關(guān)物質(zhì)分析方法開發(fā)的任務(wù)時(shí)�����,首先要對(duì)反應(yīng)線路和工藝進(jìn)行了解�����,并與合成研究員共同探討可能的工藝雜質(zhì)和降解雜質(zhì),目的是要確認(rèn)在所要開發(fā)的方法中�����,需要考慮的雜質(zhì)有哪些?哪些雜質(zhì)是需要重點(diǎn)考察(需達(dá)一定的線性�����、精密度、準(zhǔn)確度),哪些雜質(zhì)僅需滿足分離度要求即可�����,在方法開發(fā)之前需要對(duì)這些信息有大致的判斷。
Step 2 理化性質(zhì)研究
在確認(rèn)了需要控制的雜質(zhì)后�����,要對(duì)API的理化性質(zhì)進(jìn)行研究�����,例如解離常數(shù)對(duì)流動(dòng)相pH設(shè)置有提示作用�����;分配系數(shù)可以對(duì)分析系統(tǒng)和保留時(shí)間有預(yù)判�����;結(jié)構(gòu)中是否存在共軛結(jié)構(gòu)等有助于檢測(cè)器類型的選擇�����。
Step 3分析系統(tǒng)選擇
分析系統(tǒng)的選擇取決于2方面�����,一是待分析化合物的極性特征�����,二是所選擇色譜柱填料的類型�����。絕大多數(shù)情況下�����,都可以采用反相液相進(jìn)行�����。當(dāng)待測(cè)化合物極性較大且通過(guò)調(diào)整有機(jī)相比例�����、添加離子對(duì)試劑或者更換Hilic色譜柱都無(wú)法得到較好的分離效果時(shí)可以考慮正相色譜法�����,另外如果需要分析異構(gòu)體雜質(zhì)�����,并且所選擇的手性柱為正相填料�����,那么也應(yīng)該選擇正相色譜法。
Step 4 初始方法建立
在分析系統(tǒng)確定后�����,就可以建立初始分析方法了。首選有機(jī)相與水作為流動(dòng)相�����,采用最簡(jiǎn)單的勻速線性梯度,考察主峰及各雜質(zhì)的分布情況�����,對(duì)方法有機(jī)相的初始和終點(diǎn)比例有大致的判斷�����,同時(shí)對(duì)樣品進(jìn)行多波長(zhǎng)掃描,選擇可以同時(shí)兼顧主成分和雜質(zhì)分析的波長(zhǎng)。如果在有機(jī)相和水作為流動(dòng)相時(shí)化合物峰型極差可以考慮將水更換為酸溶液或緩沖鹽�����。
Step 5 方法優(yōu)化
通常初始方法的性能不能達(dá)到我們所設(shè)定的要求�����,需要繼續(xù)進(jìn)行優(yōu)化�����,主要是優(yōu)化分離度�����。分離度計(jì)算公式為:
通過(guò)公式可知,分離度是理論塔板數(shù)、選擇性和保留因子的公式�����,凡是能影響理論塔板數(shù)�����、選擇性和保留因子的因素都是方法優(yōu)化的對(duì)象�����,包括色譜柱、流動(dòng)相種類�����、pH、添加劑種類和濃度、流動(dòng)相梯度�����、柱溫等�����。
一般情況下�����,首先優(yōu)化無(wú)法量化的色譜柱和流動(dòng)相種類,然后再對(duì)流動(dòng)相pH�����、添加劑種類與濃度�����、流動(dòng)相梯度、柱溫等條件進(jìn)行優(yōu)化。
5.1 色譜柱
1)色譜柱是液相系統(tǒng)的核心,更換色譜柱填料類型(圖2)對(duì)分離度產(chǎn)生的影響是最大的。
反相色譜柱常用的填料包括C18�����、C8�����、C4、氨基柱�����、苯基柱等�����,正相色譜柱常用的填料包括多糖或淀粉鍵合柱�����,氨基柱等。填料選擇取決于待分析化合物的極性特征以及是否含有特定基團(tuán)�����,例如非極性及弱極性化合物適用于C18 色譜柱�����,中等極性化合物可使用氨基柱�����,極性化合物可嘗試Hilic色譜柱或正相色譜柱�����,芳香族化合物可以嘗試苯基柱�����。
除了填料類型�����,碳載量和比表面積也是影響保留的重要因素�����,碳載量高的色譜柱保留強(qiáng)�����,如果篩選填料類型后分離仍不理想可以考慮嘗試碳載量更高的色譜柱,也許會(huì)有較好的效果�����。
在色譜柱篩選時(shí)應(yīng)關(guān)注不同批次填料的差異�����,這將影響常規(guī)測(cè)樣期間更換色譜柱后檢測(cè)結(jié)果的重現(xiàn)性�����,嚴(yán)重時(shí)甚至可能會(huì)導(dǎo)致方法終止�����。
圖2 色譜柱填料類型
2)色譜柱粒徑和內(nèi)徑越小峰型越尖銳,容易獲得更好的分離度�����,但同時(shí)色譜柱背壓也會(huì)越高�����,在選擇這些參數(shù)時(shí)要考慮儀器的壓力耐受范圍。
3)柱長(zhǎng)與分離度成正比�����,色譜柱越長(zhǎng)�����,獲得的理論塔板數(shù)越高�����,分離越好,但待分析物保留時(shí)間會(huì)相應(yīng)增加,分析時(shí)間可能會(huì)延長(zhǎng)�����。
5.2 溶劑種類
溶劑類型會(huì)影響選擇性�����。反相色譜中常用的有機(jī)相為乙腈�����、甲醇�����,有時(shí)也會(huì)加入適量的THF以增加選擇性�����,添加量一般不超過(guò)10%�����,否則可能會(huì)耗損液相色譜儀。其中乙腈的洗脫能力更強(qiáng)�����,且截止波長(zhǎng)短�����,粘度低�����,惰性好�����,是有機(jī)相的首選�����。甲醇與乙腈洗脫能力不同�����,與待測(cè)組分及固定相的相互作用方式也不同�����,因此待測(cè)組分在甲醇中與在乙腈中可能呈現(xiàn)不同的洗脫順序或分離效果,必要時(shí)可以將乙腈-甲醇以一定比例混合作為有機(jī)相�����。
5.3 流動(dòng)相pH
目前大多數(shù)藥物具有可電離基團(tuán)�����,流動(dòng)相pH可明顯影響待分析化合物及相關(guān)雜質(zhì)的解離狀態(tài),酸性化合物在酸性條件下具有更好的峰型和保留�����,堿性化合物在堿性條件下具有更好的峰型和保留�,在pH優(yōu)化過(guò)程中除了要考慮待分析物的結(jié)構(gòu)特征外,要重點(diǎn)關(guān)注色譜柱所能耐受的使用范圍��,大多數(shù)色譜柱填料都是硅膠基質(zhì)的�,即便鍵合了惰性基團(tuán),堿性條件下也很容易溶解,因此使用高pH時(shí)一定要做詳細(xì)的技術(shù)咨詢����。
5.4 流動(dòng)相添加劑種類及濃度
1)流動(dòng)相添加劑種類包括酸和緩沖鹽,采用哪種添加劑需要通過(guò)試驗(yàn)結(jié)果來(lái)選擇����,通常情況下緩沖鹽使用更普遍��。
2)添加劑濃度可能影響待測(cè)組分的峰型和保留����,一般從低濃度開始,逐步增加濃度��,當(dāng)峰型和分離不再改善時(shí)停止優(yōu)化��,以獲得可接受分離效果所需的最低濃度作為添加劑濃度。
5.5 梯度
流動(dòng)相梯度變化可改變?nèi)軇?qiáng)度��,是改善分離的重要手段。梯度設(shè)置可依據(jù)樣品中雜質(zhì)分布情況來(lái)調(diào)節(jié)��,通常分布密集的地方可以采用更低的變化速率,雜質(zhì)稀疏的地方可以采用更快的變化速率����,但也需要注意由于不同組分對(duì)有機(jī)相變化的敏感程度不同��,也不是一味降低梯度變化速率就能獲得更好分離����,有時(shí)可能獲得相反的結(jié)果。建議在梯度篩選的開始時(shí)多設(shè)置不同的變化速率,整體考察各待測(cè)組分對(duì)梯度的敏感程度�,總結(jié)出規(guī)律,再有針對(duì)性的設(shè)置梯度��。
5.6 柱溫
隨著液相色譜的普及,柱溫作為改善分離的輔助手段的重要性越來(lái)越受到重視����。柱溫通過(guò)兩種方式影響分離度��,一是高溫可使峰型更尖銳��,從而改善分離度����;二是高溫可使各待測(cè)組分保留時(shí)間提前��,且不同的組分對(duì)溫度敏感程度不一樣(有研究表明溫度對(duì)可電離化合物的影響高于不可電離化合物)��,從而改變分離度��,但是分離度可能變好也可能變更差����,需通過(guò)試驗(yàn)數(shù)據(jù)進(jìn)行確認(rèn)��。此外����,由于高柱溫可使保留時(shí)間更短,提高分離速度�,因此可以縮短分析時(shí)間,提高工作效率�。
Step 6 方法預(yù)驗(yàn)證
分析方法開發(fā)完成后����,需要對(duì)所開發(fā)的方法進(jìn)行簡(jiǎn)單的預(yù)驗(yàn)證��,以便為后續(xù)方法學(xué)驗(yàn)證提供一個(gè)較好的輸入����。一般情況下可以對(duì)專屬性(降解試驗(yàn))、定量限����、檢測(cè)限��、回收率�、溶液穩(wěn)定性等做初步考察��,同時(shí)結(jié)合方法開發(fā)過(guò)程中對(duì)方法特性的理解��,對(duì)可能影響方法性能的條件進(jìn)行適當(dāng)?shù)哪陀眯钥疾臁?/span>
結(jié)束語(yǔ)
有關(guān)物質(zhì)方法開發(fā)的總體原則是在保證專屬性、靈敏度��、準(zhǔn)確度等必要條件下��,盡量縮短分析時(shí)間����,以提高檢測(cè)速率,同時(shí)實(shí)現(xiàn)方法的經(jīng)濟(jì)性��。
方法開發(fā)是在工藝未完全確認(rèn)的階段開展的,當(dāng)工藝發(fā)生變更時(shí)要仔細(xì)評(píng)估��,隨時(shí)調(diào)整方法��,確保方法能適用于雜質(zhì)譜研究,為后續(xù)安全性評(píng)價(jià)奠定基礎(chǔ)�。
分析方法開發(fā)沒有絕對(duì)的終點(diǎn),是否終止開發(fā)取決于方法目標(biāo)的設(shè)定��,能達(dá)到設(shè)定的目標(biāo)即可��,無(wú)需花費(fèi)更多的時(shí)間尋找更優(yōu)的方法�,因?yàn)楫?dāng)你這樣做的時(shí)候你會(huì)發(fā)現(xiàn)方法開發(fā)的工作將永遠(yuǎn)無(wú)法結(jié)束,所以不要戀戰(zhàn)��。
一旦分析方法建立并經(jīng)過(guò)驗(yàn)證,就要建立基于風(fēng)險(xiǎn)評(píng)估和可用數(shù)據(jù)的控制策略��,并在日常檢樣中持續(xù)監(jiān)控方法性能��,必要時(shí)進(jìn)行方法改進(jìn)。
參考文獻(xiàn)
[1]N.S. Wilson��,C olumn selectivity in reversed-phase liquid chromatography I. A general quantitative relationship,Journal of Chromatography A, 961 (2002) 171–193
[2]John W. Dolan�,Temperature selectivity in reversed-phase high performance liquid chromatography,Journal of Chromatography A, 965 (2002) 195–205
[3]T.L. Chester,Business-objective-directed, constraint-based multivariate optimization of high-performance liquid chromatography operational parameters,Journal of Chromatography A, 1016 (2003) 181–193
[4]M.C. Garc´?a-A´ lvarez-Coque,Models and objective functions for the optimisation of selectivity in reversed-phase liquid chromatography��,Analytica Chimica Acta 579 (2006) 125–145
[5]Dispas A, Quality by design approach for the analysis of impurities in pharmaceutical drug products and drug substances ,Tr AC-Trends Anal Chem, 2018, 101: 24-33