摘要
目的:對(duì)美國(guó)食品和藥物管理局(FDA)發(fā)布的藥物非臨床研究警告信進(jìn)行分析�����,為藥物非臨床研究質(zhì)量管理規(guī)范(GLP)監(jiān)管提供參考�����。
方法:收集2008年至2022年FDA對(duì)非臨床研究發(fā)出的警告信�,統(tǒng)計(jì)警告信數(shù)量及問(wèn)題分布情況���,解析和歸納警告信中公布的研究者在非臨床研究中發(fā)生的主要問(wèn)題�。
結(jié)果:FDA對(duì)非臨床研究發(fā)布的12封警告信中共有67項(xiàng)問(wèn)題數(shù)�,涉及21CFR58條款120項(xiàng)次�����。其中發(fā)生頻率最高的缺陷是質(zhì)量保證部門人員履職不足���、專題負(fù)責(zé)人履職不足和研究過(guò)程中方案的實(shí)施及報(bào)告撰寫。
結(jié)論:FDA對(duì)藥物非臨床研究警告信中高頻率問(wèn)題集中于項(xiàng)目負(fù)責(zé)人及質(zhì)量保證部門的履職盡責(zé)方面。監(jiān)管機(jī)構(gòu)應(yīng)在人員的履職盡責(zé)方面重點(diǎn)關(guān)注���,促進(jìn)非臨床研究質(zhì)量的提升�。
關(guān)鍵詞
藥物非臨床研究質(zhì)量管理規(guī)范;警告信�����;美國(guó)食品和藥物管理局�����;研究人員
藥物非臨床研究是為評(píng)價(jià)藥物安全性�,在實(shí)驗(yàn)室條件下用實(shí)驗(yàn)系統(tǒng)進(jìn)行的實(shí)驗(yàn),是藥物研發(fā)的基礎(chǔ)工作,也是臨床試驗(yàn)的重要基礎(chǔ)數(shù)據(jù)���,被認(rèn)為是人體試驗(yàn)前的最后一道防線[1]�����。藥物非臨床研究質(zhì)量管理規(guī)范(non-clinical good laboratory practice�,GLP)是藥物非臨床研究的質(zhì)量管理和保證體系[2]�。美國(guó)食品和藥物管理局(FDA)于1979年實(shí)施了美國(guó)聯(lián)邦法規(guī)21CFR58《GLP規(guī)范(草案)》[3],主要適用于FDA產(chǎn)品研究或市場(chǎng)許可而進(jìn)行的非臨床研究�����,其產(chǎn)品主要包括人用藥和動(dòng)物用藥、生物制品���、人用醫(yī)療器械、電子產(chǎn)品�����、食品和顏色添加劑以及飼料添加劑�。我國(guó)《藥物非臨床研究質(zhì)量管理規(guī)范》于2006年正式實(shí)施�,2017年進(jìn)行了修訂[4]���。
FDA要求開展安全性評(píng)價(jià)研究的非臨床實(shí)驗(yàn)室符合GLP要求�����,保證安全性數(shù)據(jù)的質(zhì)量和可靠性。FDA的GLP檢查包括監(jiān)督檢查和定向檢查���,監(jiān)督檢查包括定期檢查和常規(guī)性檢查,定向檢查包括注冊(cè)核查�����、有因檢查�����、跟蹤檢查和整改檢查[5]���。GLP常規(guī)檢查每2年產(chǎn)生一次計(jì)劃,其他類型檢查按照需要安排時(shí)間�。FDA警告信是FDA針對(duì)現(xiàn)場(chǎng)檢查中發(fā)現(xiàn)的顯著違規(guī)行為對(duì)檢查對(duì)象發(fā)出的警戒,同時(shí)在FDA網(wǎng)站上對(duì)外公布���,是監(jiān)管過(guò)程中主要監(jiān)管措施[6]���。本研究針對(duì)FDA在現(xiàn)場(chǎng)檢查中針對(duì)研究者發(fā)布的警告信進(jìn)行分析,對(duì)涉及的法規(guī)及條款進(jìn)行歸納整理分析���,以期為我國(guó)藥物非臨床安全性評(píng)價(jià)研究提供參考�����。
資料與方法
數(shù)據(jù)來(lái)源
選擇2008年至2022年FDA網(wǎng)站上針對(duì)非臨床研究發(fā)出的警告信作為分析數(shù)據(jù)來(lái)源���。
分析方法
統(tǒng)計(jì)警告信的數(shù)量及提出問(wèn)題的數(shù)量�,對(duì)問(wèn)題進(jìn)行分類整理。將警告信提出的問(wèn)題分別對(duì)應(yīng)21CFR食品和藥品條款和我國(guó)GLP法規(guī)進(jìn)行分類,描述性統(tǒng)計(jì)各類問(wèn)題�。
結(jié)果
2008年至2022年期間���,F(xiàn)DA對(duì)非臨床研究發(fā)布的12封警告信[7]中共有67項(xiàng)問(wèn)題數(shù),涉及21CFR58條款120項(xiàng)次�����,見(jiàn)表1。
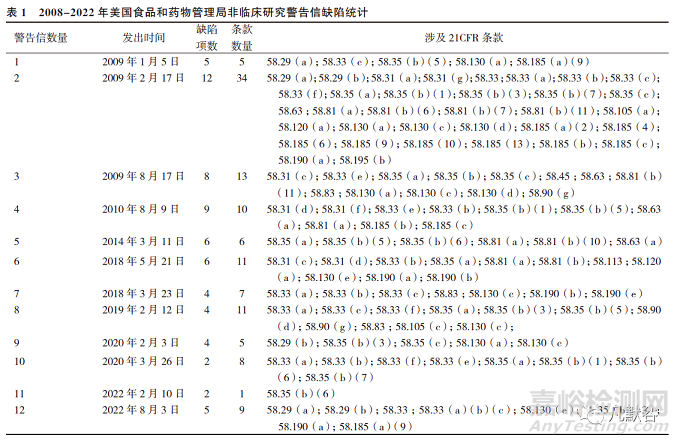
按照21CFR58的分類,警告信中發(fā)生率最高的缺陷是質(zhì)量保證人員�、專題負(fù)責(zé)人、非臨床研究方案的實(shí)施和非臨床研究結(jié)果的報(bào)告�����,分別占比21.7%���、19.2%、10.8%和10.0%�,主要表現(xiàn)在專題負(fù)責(zé)人及質(zhì)量保證人員未履行相應(yīng)的職責(zé),研究未按照方案進(jìn)行�����,報(bào)告中對(duì)數(shù)據(jù)進(jìn)行的轉(zhuǎn)換與計(jì)算缺少描述等,見(jiàn)表2�����。
討論
對(duì)2008年至2022年FDA發(fā)出警告信進(jìn)行分析統(tǒng)計(jì)�,發(fā)出的警告信數(shù)量及提出問(wèn)題的數(shù)量呈整體下降趨勢(shì),表明非臨床實(shí)驗(yàn)質(zhì)量在逐年上升�。2008年至2018年期間FDA發(fā)出的警告信中多次出現(xiàn)標(biāo)準(zhǔn)操作規(guī)程不完善不合理的問(wèn)題,而在2019年及以后未再出現(xiàn)�。質(zhì)量保證人員履職不足、專題負(fù)責(zé)人未確保研究按照方案進(jìn)行的問(wèn)題長(zhǎng)期存在�,是影響非臨床實(shí)驗(yàn)質(zhì)量的重要因素。
2021年FDA未發(fā)出GLP警告信�,根據(jù)FDA2021年GLP現(xiàn)場(chǎng)檢查[8]數(shù)據(jù)統(tǒng)計(jì)�����,現(xiàn)場(chǎng)檢查發(fā)現(xiàn)缺陷主要是儀器設(shè)備管理���、培訓(xùn)和專題負(fù)責(zé)人三個(gè)方面�����,涉及最多的條款是21CFR58.130(a)�����,主要體現(xiàn)在未確保儀器設(shè)備使用人員掌握儀器所具備的功能�����;未確保設(shè)施所在工作環(huán)境條件是否可滿足需要;未確保設(shè)施是否運(yùn)轉(zhuǎn)正常���,是否存在會(huì)對(duì)研究結(jié)果造成干擾的因素;儀器使用的培訓(xùn)未能覆蓋所有儀器使用人員�����;專題負(fù)責(zé)人未能確保研究按照方案進(jìn)行���;實(shí)驗(yàn)方案變更未能及時(shí)進(jìn)行記錄���;未能及時(shí)�����、準(zhǔn)確�����、完整地記錄原始數(shù)據(jù)等�。警告信所提問(wèn)題與FDA在核查過(guò)程中發(fā)現(xiàn)問(wèn)題的總體情況可能存在一定差異���。警告信中所提的問(wèn)題僅是針對(duì)顯著違規(guī)行為���,若違規(guī)者不及時(shí)、不充分依據(jù)警告信的要求糾正其違規(guī)行為�����,可能會(huì)導(dǎo)致FDA向法院提起執(zhí)行訴訟�����。因此���,對(duì)警告信內(nèi)容進(jìn)行系統(tǒng)分析�����,可針對(duì)性地對(duì)監(jiān)管模式進(jìn)行優(yōu)化,實(shí)現(xiàn)更有效的監(jiān)管�,保證公共用藥安全。對(duì)于一般性的違規(guī)行為�,F(xiàn)DA通常會(huì)向違規(guī)者發(fā)出無(wú)標(biāo)題信,并在FDA網(wǎng)站上公布�����,但不公布違規(guī)者的相關(guān)信息[9]�����。
GLP認(rèn)證[10]是指國(guó)家藥品監(jiān)督管理局依照申請(qǐng)組織對(duì)藥物非臨床安全性評(píng)價(jià)研究機(jī)構(gòu)進(jìn)行GLP情況的檢查�、評(píng)定的過(guò)程。與FDA基于風(fēng)險(xiǎn)�,針對(duì)項(xiàng)目的檢查不同,我國(guó)重點(diǎn)是對(duì)實(shí)驗(yàn)機(jī)構(gòu)進(jìn)行檢查���。機(jī)構(gòu)只有通過(guò)認(rèn)證檢查后���,才可以開展相關(guān)研究�。2022年�,國(guó)家藥品監(jiān)督管理局食品藥品審核查驗(yàn)中心(CFDI)發(fā)布的《2021年度藥品檢查工作報(bào)告》[11]顯示,2021年GLP認(rèn)證檢查任務(wù)38個(gè)�,不通過(guò)的任務(wù)1個(gè)。GLP認(rèn)證檢查發(fā)現(xiàn)的主要問(wèn)題包括:質(zhì)量保證部門履職能力不足�;機(jī)構(gòu)負(fù)責(zé)人資質(zhì)不符合要求;實(shí)驗(yàn)動(dòng)物設(shè)施設(shè)計(jì)與管理不到位�����;室驗(yàn)環(huán)節(jié)未能嚴(yán)格執(zhí)行操作規(guī)程�����;計(jì)算機(jī)賬戶權(quán)限分級(jí)設(shè)置不合理等���。國(guó)家藥品監(jiān)督管理局于2023年1月19日發(fā)布《藥物非臨床研究質(zhì)量管理規(guī)范認(rèn)證管理辦法》[12]�����,進(jìn)一步規(guī)范GLP認(rèn)證和監(jiān)督管理工作�����。
本研究歸納分析2008年至2022年FDA針對(duì)GLP警告信�,發(fā)現(xiàn)質(zhì)量保證人員履職不足發(fā)生頻次最高�����,占比21.7%�,這同樣是CFDI在2021年GLP檢查中發(fā)現(xiàn)的主要問(wèn)題�����。質(zhì)量保證部門是GLP體系中重要的一環(huán)�����,通過(guò)質(zhì)量保證人員的檢查,確保整個(gè)實(shí)驗(yàn)儀器設(shè)施�����、人員資質(zhì)及培訓(xùn)�、實(shí)驗(yàn)方法、操作�����、記錄等均嚴(yán)格遵守GLP規(guī)范�����。FDA與我國(guó)法規(guī)均對(duì)質(zhì)量保證部門提出對(duì)非臨床研究進(jìn)行檢查并將檢查結(jié)果匯報(bào)給專題負(fù)責(zé)人的要求�。質(zhì)量保證部門應(yīng)當(dāng)確保足夠的時(shí)間間隔檢查每項(xiàng)非臨床研究�,并檢查每次實(shí)驗(yàn)的書面記錄和正確簽名記錄,確保實(shí)驗(yàn)結(jié)果的可靠�����、準(zhǔn)確�、完整�、可追溯���。質(zhì)量保證部門的檢查貫穿于整個(gè)實(shí)驗(yàn)���,從方案的審查、實(shí)驗(yàn)的進(jìn)行�、報(bào)告審核,還有人員���、儀器���、標(biāo)準(zhǔn)操作規(guī)程(SOP)�����、原始記錄等���,若對(duì)每一項(xiàng)的所有環(huán)節(jié)與內(nèi)容進(jìn)行檢查�����,可能會(huì)由于時(shí)間或環(huán)境條件的限制���,無(wú)法進(jìn)行深入的檢查,缺乏對(duì)高風(fēng)險(xiǎn)點(diǎn)的關(guān)注�。各實(shí)驗(yàn)機(jī)構(gòu)應(yīng)當(dāng)根據(jù)自身的情況,建立具有自身特點(diǎn)的檢查方式�,并根據(jù)情況變化做出及時(shí)調(diào)整。FDA在2022年給Toxikon Corporation/ Labcorp Bedford LLC的警告信[13]中指出�,質(zhì)量保證部門未能審核研究報(bào)告�����,無(wú)法確保該報(bào)告準(zhǔn)確描述了實(shí)驗(yàn)方法�,無(wú)法確保實(shí)驗(yàn)結(jié)果準(zhǔn)確反映出實(shí)驗(yàn)的原始數(shù)據(jù)���。實(shí)驗(yàn)重要指標(biāo)數(shù)據(jù)血漿體積計(jì)算錯(cuò)誤產(chǎn)生計(jì)算誤差,導(dǎo)致隨后的腎血漿流量�����、腎血流量和腎小球?yàn)V過(guò)率均產(chǎn)生計(jì)算誤差���,且未能糾正1h時(shí)間點(diǎn)時(shí)尿量計(jì)算問(wèn)題。2021年10月6日質(zhì)量保證部門審查后�,并未發(fā)現(xiàn)此問(wèn)題。2021年10月14日的修訂報(bào)告未能糾正所有偏差問(wèn)題�。
警告信中提出報(bào)告中缺少使用的基本計(jì)算和公式。Toxikon Corporation/Labcorp Bedford LLC與FDA均無(wú)法保證最終研究報(bào)告中的內(nèi)容是準(zhǔn)確的���,質(zhì)量保證工作的失職使整個(gè)研究的有效性受到質(zhì)疑。警告信中出現(xiàn)頻率第二的問(wèn)題是專題負(fù)責(zé)人�����,同時(shí)也是2021年FDA現(xiàn)場(chǎng)檢查總結(jié)報(bào)告中的高發(fā)問(wèn)題���,此處的專題負(fù)責(zé)人與我國(guó)的專題負(fù)責(zé)人職責(zé)不完全相同。FDA的專題負(fù)責(zé)人除了對(duì)研究的執(zhí)行負(fù)責(zé)�����,還要審查批準(zhǔn)實(shí)驗(yàn)方案及總結(jié)報(bào)告。而我國(guó)的實(shí)驗(yàn)方案及總結(jié)方案由機(jī)構(gòu)負(fù)責(zé)人批準(zhǔn)���,專題負(fù)責(zé)人在研究中應(yīng)當(dāng)確保研究遵循研究方案,確保所有實(shí)驗(yàn)數(shù)據(jù)均準(zhǔn)確記錄并進(jìn)行驗(yàn)證���,確保在研究期間或研究結(jié)束時(shí)將原始數(shù)據(jù)���、文件、方案�����、樣本和最終報(bào)告存檔���,確保遵守所有GLP規(guī)定�。FDA在2020年給University of Kentucky的警告信[14]中指出其未能遵守研究方案進(jìn)行研究���,無(wú)法確保數(shù)據(jù)的質(zhì)量和可靠性。此外�,未能保留和記錄原始數(shù)據(jù)和樣本會(huì)妨礙對(duì)研究結(jié)果的審查、分析和驗(yàn)證���。確保完整和準(zhǔn)確的研究數(shù)據(jù)�����,F(xiàn)DA才能在臨床試驗(yàn)開始之前充分評(píng)估該研究的總體安全性和風(fēng)險(xiǎn)�。專題負(fù)責(zé)人除了需要有勝任資格�����,還應(yīng)加強(qiáng)對(duì)研究工作人員的培訓(xùn)與考核���。2021年FDA發(fā)出的檢查報(bào)告中也頻繁提及:所有研究工作人員都應(yīng)在研究開始前進(jìn)行GLP培訓(xùn)�,規(guī)避相關(guān)問(wèn)題的發(fā)生。
非臨床研究方案的實(shí)施和非臨床研究結(jié)果的報(bào)告也是FDA警告信中提及較多的問(wèn)題。FDA在2022年給Valley Biosystems的警告信[15]中提出所有實(shí)驗(yàn)數(shù)據(jù)���,包括測(cè)試系統(tǒng)意外響應(yīng)的觀察結(jié)果,均需要準(zhǔn)確記錄并驗(yàn)證�����;出現(xiàn)可能會(huì)影響非臨床研究質(zhì)量的情況時(shí)應(yīng)謹(jǐn)慎對(duì)待���,采取糾正措施并記錄在案���;修訂相關(guān)SOP,防止此類偏差在未來(lái)GLP研究中再次出現(xiàn)�����,并需提交新的或修訂的標(biāo)準(zhǔn)操作規(guī)程的相關(guān)培訓(xùn)文件。
綜上���,F(xiàn)DA對(duì)于非臨床研究警告信中提及頻率最高的問(wèn)題集中于質(zhì)量保證部門、專題負(fù)責(zé)人的履職盡責(zé)和非臨床研究方案的實(shí)施及報(bào)告���。一個(gè)可靠的質(zhì)量保證部門是GLP研究成功不可或缺的,最終報(bào)告中細(xì)微的誤差可能會(huì)對(duì)結(jié)果產(chǎn)生巨大影響�;專題負(fù)責(zé)人的規(guī)范操作直接影響到研究的質(zhì)量�����;實(shí)驗(yàn)過(guò)程應(yīng)嚴(yán)格按照方案實(shí)施�。GLP體系的建立,有助于把控研究的每一個(gè)環(huán)節(jié)�,降低誤差,確保結(jié)果的真實(shí)與準(zhǔn)確�����。本研究提示,監(jiān)管機(jī)構(gòu)應(yīng)在落實(shí)責(zé)任�����、規(guī)范操作���、加強(qiáng)培訓(xùn)幾個(gè)方面重點(diǎn)關(guān)注專題負(fù)責(zé)人及質(zhì)量保證部門,確保實(shí)驗(yàn)數(shù)據(jù)的真實(shí)可靠�,避免發(fā)生嚴(yán)重違規(guī)行為���,促進(jìn)非臨床研究質(zhì)量的提高。


