植入式醫(yī)療器械通常是高風(fēng)險醫(yī)療器械����,其潛在故障可能會給患者帶來嚴重后果��。這就是監(jiān)管機構(gòu)高度關(guān)注此類醫(yī)療器械安全性的原因——特別是在新歐盟醫(yī)療器械法規(guī)2017/745中��。
在本文中�����,我們將根據(jù)歐盟法規(guī)介紹與植入式醫(yī)療器械相關(guān)的主要要求。
鑒于這些類型設(shè)備的特殊性�����,有一些特定要求需要考慮����,并應(yīng)在醫(yī)療器械文件或其他類型的文件中提供合規(guī)性證據(jù),例如在法規(guī)遵從策略背景下��。
1��、什么是植入式醫(yī)療器械��?
歐盟MDR文本中規(guī)定的植入式醫(yī)療器械定義如下:
“植入式器械”是指部分或完全吸附的醫(yī)療器械����,其目的是:完全進入人體或借助臨床手術(shù)替代人體上皮表面或眼表面,并且在手術(shù)后保留在人體內(nèi)�����。
任何借助臨床手術(shù)部分進入人體內(nèi)��,并且在手術(shù)后留在人體內(nèi)至少30天的醫(yī)療器械也應(yīng)被視為植入式器械����。
2、植入式器械的一般考慮因素
與其他醫(yī)療器械一樣�����,植入式醫(yī)療器械可以分為有源器械和無源器械�����。無源器械內(nèi)部沒有能量源�����,并且可以通過其他方式激活�����,如患者的運動或呼吸����。
而有源植入式醫(yī)療器械是通過自然腔道(口)或手術(shù)方式插入患者體內(nèi)的通電設(shè)備,并在手術(shù)后保留在患者體內(nèi)��。
有源植入式醫(yī)療器械被認為是高風(fēng)險器械����,需要實現(xiàn)高標準制造和更高水平的法規(guī)合規(guī)性����。
3��、ISO 13485要求
ISO 13485:2016包含與植入式醫(yī)療器械相關(guān)的多項要求�����,匯總?cè)缦聢D所示����。我們將首先了解主要要求,然后再轉(zhuǎn)向歐盟MDR 2017/745中的規(guī)定要求��。
植入式醫(yī)療器械
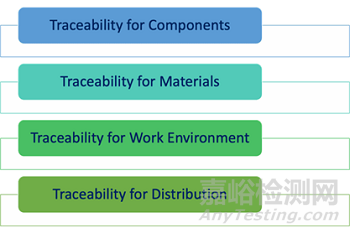
第7.5.9.2節(jié)規(guī)定了可追溯性方面的特定要求����。具體而言,應(yīng)保留以下方面的可追溯性記錄:
部件��。重要的是����,如果醫(yī)療器械由不同的部件成,則應(yīng)保持所有這些部件的可追溯性����。基本上����,每個部件都應(yīng)與特定BOM和特定的生產(chǎn)記錄關(guān)聯(lián)。如此����,在任何時候都可以追溯到指定醫(yī)療器械中包含的部件。
材料��。用于制造植入式醫(yī)療器械的所有材料都應(yīng)被正確識別和追蹤�����;應(yīng)提供材料適合用于此類醫(yī)療器械的具體證明��,因為這對于患者的整體安全至關(guān)重要����。
工作環(huán)境。顯而易見的是工作環(huán)境對于植入式醫(yī)療器械的影響是巨大的,特定工作條件可能會直接影響器械的質(zhì)量和安全��。因此����,有必要準確記錄工作環(huán)境條件,如溫度��、濕度等����。
此外,植入式醫(yī)療器械的可追溯性附加要求如下:
機構(gòu)/企業(yè)應(yīng)要求分銷服務(wù)供應(yīng)商或分銷商保存醫(yī)療器械的分銷記錄��,以實現(xiàn)可追溯性����,并確保這些記錄可供檢查。
這與分銷服務(wù)供應(yīng)商或分銷商有關(guān)��,他們有義務(wù)保存醫(yī)療器械的分銷記錄����。這點至關(guān)重要,因為始終有必要了解最終患者�����,以便在發(fā)現(xiàn)任何安全問題時可以與患者快速聯(lián)系。
最后����,ISO 13485:2016第8.2.6節(jié)中還提出了對植入式醫(yī)療器械的另一項要求�����,其中明確規(guī)定必須記錄在產(chǎn)品發(fā)布過程中進行醫(yī)療器械檢查或測試的所有人員的身份��。
Medtec China已經(jīng)從2012年開始連續(xù)舉辦了8屆植入介入醫(yī)療器械峰會�����,會議圍繞骨科植入物����、心血管介入產(chǎn)品,探討其法規(guī)政策����、市場趨勢、研發(fā)與設(shè)計與材料創(chuàng)新等內(nèi)容����,為植入介入制造商材料供應(yīng)商及服務(wù)商等提供一個講解公司新技術(shù)及材料的平臺����。掃描下方二維碼立刻報名參展加入我們~
4����、歐盟MDR 107/745要求
根據(jù)EU MDR 2017/745,與植入式醫(yī)療器械相關(guān)的最重要的新要求是所謂的植入卡����。該要求旨在改善與患者共享的植入式醫(yī)療器械的相關(guān)信息。
植入卡有不同的用途��,例如:
使患者能夠識別植入的醫(yī)療器械并獲取安全相關(guān)信息����。
使患者能夠在特定情況下識別自己是需要特殊護理的人員,例如安全檢查或其他情況����。
該法規(guī)第18條定義了植入卡的相關(guān)要求,其中明確規(guī)定了植入卡中需要包含的信息����。具體而言��,應(yīng)包括以下信息:
器械名稱
器械類型
唯一器械標識(UDI)– DI和PI均應(yīng)包含在內(nèi)
序列號或批號(如適用)
醫(yī)療器械制造商的名稱和地址
醫(yī)療器械制造商的網(wǎng)站
患者或醫(yī)療專業(yè)人員針對可合理預(yù)見的外部影響����、醫(yī)療檢查或環(huán)境條件的相互干擾����,所應(yīng)采取的任何警告����、預(yù)防措施或糾正措施,如第18(b)條所述��。
醫(yī)療器械的預(yù)期使用壽命
確?���;颊甙踩褂闷餍档钠渌畔?/span>
此外,還需要包含患者的信息�����。包括患者姓名��、植入日期和進行器械植入的醫(yī)療機構(gòu)��。這些信息應(yīng)在植入卡交給患者時就包含在內(nèi),以便盡可能地尊重隱私和GDPR要求��。
此外��,植入卡還有其他具體要求����,如下圖中所述:
我們將不會詳細介紹所有這些具體要求,但很重要的一點是����,MDCG(歐盟醫(yī)療器械協(xié)調(diào)小組)發(fā)布了一份具體指南MDCG 2019-08,其中說明了與植入卡相關(guān)的所有要求��。
5��、植入式醫(yī)療器械相關(guān)的ISO標準
在產(chǎn)品設(shè)計����、生產(chǎn)和后期制造階段,還需要適當考慮與植入式醫(yī)療器械相關(guān)的其他標準��。下面是我們找到的與植入式醫(yī)療器械相關(guān)的最重要的ISO標準清單����;但這并不是詳盡清單��,根據(jù)設(shè)備的類型��,其他ISO標準也可能適用��。
ISO 14708-1:2014 – 安全�����、標記和制造商所提供信息的通用要求
ISO 14708-2:2012 – 心臟起搏器
ISO 14708-3:2017 – 植入式神經(jīng)刺激器
ISO 14708-4:2008 – 植入式輸液泵
ISO 14708-5:2010 – 循環(huán)支持器械
ISO 14708-6:2010 – 治療快速心律失常的有源植入式醫(yī)療器械的特殊要求(包括植入式心臟除顫器)
ISO 14708-7:2013 – 人工耳蝸系統(tǒng)的特殊要求
ISO 14117:2012 – 植入式心臟起搏器��、植入式心臟復(fù)律除顫器和心臟再同步器械的電磁兼容性測試協(xié)議
ISO 14602:2010 – 無源外科植入物 – 骨接合植入物 – 特殊要求
ISO 14630:2012 – 無源外科植入物 – 通用要求
EC 62304:2006 – 醫(yī)療器械軟件
6、技術(shù)文件的合規(guī)性檢查清單
為了便于在向公告機構(gòu)提交檔案之前控制技術(shù)文件的合規(guī)性��,QualityMedDev根據(jù)MDR 2017/745編制了一份合規(guī)性檢查清單�����,其中詳細列出了對技術(shù)文件的所有要求��。
該技術(shù)文件檢查清單將作為簡化技術(shù)檔案符合性評估的重要工具����。這份15頁的檢查清單以Word文件形式提供,因此是完全可編輯的��,可適應(yīng)貴組織推向市場的產(chǎn)品類型。
翻譯文章來源:QualityMedDev