[摘要]注射劑中的可見異物直接關(guān)系到患者用藥安全,是注射劑一項重要的質(zhì)量指標���,近年來可見異物檢查不合格以及由此引起的注射劑召回事件頻發(fā)�。因此�,注射劑中可見異物的風(fēng)險評估、生產(chǎn)過程控制和產(chǎn)品檢驗應(yīng)引起高度重視����。本文參考美國FDA發(fā)布的《注射劑可見異物檢查行業(yè)指南(草案)》,同時結(jié)合國內(nèi)外相關(guān)指南和審評工作��,從可見異物概念�、風(fēng)險評估、生產(chǎn)過程控制和樣品檢查�����、生命周期管理等方面對注射劑可見異物的控制策略進行介紹��,以期為我國注射劑可見異物的控制和評價提供參考�����。
注射劑中的可見異物系指在規(guī)定條件下目視可以觀測到的不溶性物質(zhì)��,其粒徑或長度通常 > 50 μm[1]。注射劑中存在的可見異物進入體內(nèi)可引起血管栓塞�����、靜脈炎����、肉芽腫和變態(tài)反應(yīng)等�,甚至全身感染,直接危害患者用藥安全[2-4]�。國內(nèi)外注射劑中可見異物檢查不合格以及由此引起的藥品召回事件頻發(fā)[5-8],美國 FDA 發(fā)布的 2017 年 5 月—2022 年 5月近5年共344 起藥品召回事件中[9]�,涉及可見異物不合格的高達30起,占比9.0%���,僅次于微生物污染(50 起)�����。
可見異物是注射劑一項重要的質(zhì)量控制指標�����,國內(nèi)外藥品審評及監(jiān)管機構(gòu)都高度關(guān)注注射劑中可見異物的檢查和控制��。國內(nèi)外藥典均規(guī)定注射劑應(yīng)對可見異物進行檢查�,并制定了可見異物檢查方法通則,包括檢查方法和結(jié)果判定標準����。《中華人民共和國藥典》(ChP)[10]規(guī)定注射劑不得檢出明顯可見異物�,對于點狀物、2 mm 以下的短纖維和塊狀物等微細可見異物也根據(jù)不同類型藥物和不同給藥途徑制定了相應(yīng)的可接受標準��。 美國藥典(USP)[11 - 12]規(guī)定注射劑應(yīng)基本不含可見異物( essentiallyfreefromvisibleparticulates)��,歐洲藥典(EP)[13]和日本藥典(JP)[14] 對注射劑中可見異物的要求與 USP 相似�����。同時各國藥典和相關(guān)技術(shù)要求均明確注射劑生產(chǎn)過程中應(yīng)對可見異物進行 100% 檢查并同時剔除不合格產(chǎn)品��。美國 FDA 于 2021 年 12 月發(fā)布了《注射劑可見異物檢查行業(yè)指南(草案)》[15][InspectionofInjectable Products for Visible Particulates Guidancefor Industry(Draft Guidance)��,以下簡稱 FDA 指南草案]���,系統(tǒng)闡述了基于風(fēng)險評估與生命周期管理的整體可見異物檢查和控制策略�����,同時提出應(yīng)在嚴格確保注射劑生產(chǎn)符合《 藥品生產(chǎn)質(zhì)量管理規(guī)范》(GMP)要求的基礎(chǔ)上��,建立整體的控制策略�。
本文主要參考 FDA 指南草案,并結(jié)合 USP[16]�、美國注射劑協(xié)會(PDA) TR No. 79[17]���、ChP[1]等國內(nèi)外相關(guān)技術(shù)要求及實際審評工作經(jīng)驗�,從可見異物概念�����、風(fēng)險評估�����、生產(chǎn)過程控制和樣品檢查�、生命周期管理等方面對注射劑中可見異物的控制策略進行梳理介紹,以期為我國注射劑可見異物的檢查����、控制和評價提供參考。
一��、可見異物概念
可見異物是指注射劑中除氣泡以外非有意添加的、可隨液體流動���、不溶的顆粒[15����,18]��,如金屬屑����、玻璃屑、纖維�、橡膠、沉淀物等����。 FDA 指南草案未明確可見異物大小,ChP 明確指出可見異物的粒徑或長度通常 > 50 μm[1]�。
FDA 指南草案根據(jù)可見異物的來源與性質(zhì),將可見異物分為以下3 類��。
1.1 固有異物
與特定產(chǎn)品或處方相關(guān)�����,如混懸液、乳劑或其他微粒藥物遞送系統(tǒng)等[16]�����,屬于制劑目標產(chǎn)品質(zhì)量概況的一部分����。 通常情況下研究者對固有異物掌握信息相對較全面,因此其產(chǎn)生的風(fēng)險也相對較低��。
1.2 內(nèi)源性異物
是指從生產(chǎn)設(shè)備����、處方或容器密封系統(tǒng)等引入的可見異物�,一般與制劑生產(chǎn)工藝相關(guān)。 此類異物通常已隨制劑一并經(jīng)過滅菌或無菌處理����,且研究者對其來源及可能的相互作用等信息掌握相對較全面[16],但與固有異物相比仍存在一定風(fēng)險�。
1.3 外源性異物
是指從生產(chǎn)環(huán)境中引入的異物,一般屬于生產(chǎn)工藝之外的異物��,比如頭發(fā)�、與生產(chǎn)無關(guān)的纖維�����、淀粉��、礦物及類似的無機或有機材料等[16]��。外源性異物通常是一次性出現(xiàn)�����,隨機性很大�����,但由于一般無法準確獲得其來源��、進入制劑的途徑及可能的相互作用等信息��,因此可能存在微生物污染的風(fēng)險[16]����。
國內(nèi)的可見異物分類與 FDA 指南草案存在較大差異��。 ChP 大體上按不溶性物質(zhì)大小并結(jié)合材質(zhì)將可見異物分為明顯可見異物和細微可見異物�,材質(zhì)上細分為金屬類�����、玻璃類��、纖維類�、蛋白絮狀物和其他類型�����,未將來源和風(fēng)險等級等因素考慮在內(nèi)�����,同時 FDA 指南草案中所說的固有異物也不屬于 ChP可見異物的范疇�����。 相比之下��,FDA 指南草案從來源���、性質(zhì)與風(fēng)險等級等方面對可見異物進行分類,針對不同類別的可見異物分別制定相應(yīng)的控制策略����,分類更加合理科學(xué)��,更有利于可見異物控制策略的制定����。
二����、風(fēng)險評估
注射劑中的可見異物嚴重影響了藥品的質(zhì)量,患者使用后可導(dǎo)致血管栓塞����、靜脈炎、肉芽腫和變態(tài)反應(yīng)��,甚至全身感染等嚴重不良反應(yīng)事件�����,直接危害患者用藥安全����,需要嚴格預(yù)防和控制注射劑中的可見異物問題。 為確保藥品質(zhì)量并降低臨床風(fēng)險,應(yīng)在產(chǎn)品開發(fā)過程對可見異物進行風(fēng)險評估�����。 采用適當?shù)姆治黾夹g(shù)識別出典型可見異物并對其尺寸����、數(shù)量及組成成分等進行表征和鑒定,進一步追溯其潛在來源并評估風(fēng)險等級����,從而有針對性地制定可見異物控制策略。 通常固有異物�����、內(nèi)源性異物與外源性異物可能引發(fā)的風(fēng)險呈逐漸增加趨勢����,風(fēng)險降低措施也應(yīng)逐漸強化和全面。常見的可見異物分析識別技術(shù)[5�,19]通常包括顯微鏡觀察����、拉曼光譜、激光誘導(dǎo)擊穿光譜、紅外光譜等���。
2.1 固有異物
如果固有異物屬于產(chǎn)品特性且滿足制劑放行標準����,則該異物可接受�,但應(yīng)在穩(wěn)定性期間考察固有異物的尺寸或數(shù)量變化趨勢。 對于含固有異物的注射劑�,可見異物檢查存在較大的挑戰(zhàn),建議開發(fā)相關(guān)的補充檢查方法���,詳見“3. 1. 4”項特殊注射劑的考量����。
2.2 內(nèi)源性異物
針對不同來源的內(nèi)源性異物����,在制定控制策略時應(yīng)有相應(yīng)的考量。一方面����,對于可能由生產(chǎn)設(shè)備(如配液罐、生產(chǎn)管路��、硅膠管、過濾器���、墊圈等)��、原輔料或直接接觸藥包材(如玻璃或塑料瓶��、膠塞����、硅油等) 等引入的內(nèi)源性異物�,首先應(yīng)考慮從源頭控制,結(jié)合產(chǎn)品特性謹慎選擇可滿足制劑需求的原輔包��,制定比成品可接受標準更加嚴格的入廠標準�,關(guān)注清洗工藝對設(shè)備、直接接觸藥包材等引入異物或顆粒的清除能力[16]�;其次,生產(chǎn)設(shè)施設(shè)備均應(yīng)符合GMP要求����,生產(chǎn)過程須嚴格遵守 GMP 對人員、廠房與設(shè)施設(shè)備���、物料與產(chǎn)品、生產(chǎn)管理、質(zhì)量控制與質(zhì)量保證的要求��;最后�����,應(yīng)考慮由于設(shè)備磨損����、包材破損等引入異物的風(fēng)險,通過優(yōu)化操作�、維護設(shè)備等最大限度減少此類異物的引入;還可以通過定期評估特定設(shè)備生產(chǎn)不合格產(chǎn)品的趨勢�,按照生命周期管理的方式采取措施降低異物引入風(fēng)險。
另一方面�,內(nèi)源性異物也可能由制劑本身或者藥物與容器密封系統(tǒng)相互作用等引入,例如:原料藥析晶產(chǎn)生的沉淀或降解產(chǎn)生的不溶性物質(zhì)�,藥物與容器密封系統(tǒng)相互作用導(dǎo)致的玻璃脫片或侵蝕產(chǎn)生的異物,藥物與硅油或包材浸出物相互作用產(chǎn)生的異物等����。 此類內(nèi)源性異物通常在制劑放行后產(chǎn)生,且尺寸或數(shù)量可能隨放置時間而發(fā)生變化��,因此在產(chǎn)品開發(fā)階段�����,應(yīng)研究在加速或劇烈/ 強力條件下此類異物產(chǎn)生的風(fēng)險,以確定異物特征以及異物產(chǎn)生或增長與時間的相關(guān)性�,以便在穩(wěn)定性考察階段監(jiān)測該類異物的變化趨勢。 此外�����,還應(yīng)開發(fā)適用于表征和監(jiān)測該產(chǎn)品特定異物的分析方法���,通過優(yōu)化產(chǎn)品處方工藝和容器密封系統(tǒng)�,進一步減少與制劑相關(guān)的內(nèi)源性異物產(chǎn)生�����。
2.3 外源性異物
通常來源于預(yù)期不會與產(chǎn)品直接接觸的材料或環(huán)境����,一般是由于生產(chǎn)設(shè)施條件較差導(dǎo)致,如環(huán)境控制不佳��、設(shè)備老化�����、材料和人員流動等,可能存在GMP 問題并引發(fā)微生物污染�。 因此�����,注射劑必須嚴格按照 GMP 的要求組織生產(chǎn)�����,避免外源性異物的引入�����。
研究者在確定目標產(chǎn)品質(zhì)量概況以及建立可見異物的控制策略時除需考慮可見異物的類型外�����,還需考慮可見異物本身的性質(zhì)(如尺寸��、形狀����、數(shù)量、對細胞或組織的化學(xué)反應(yīng)性��、感染性、致癌性等)�����、給藥途徑�、用藥人群等臨床因素以及產(chǎn)品劑型等,例如:靜脈給藥可能比皮下注射����、肌內(nèi)注射給藥產(chǎn)生的不良事件風(fēng)險更高,對年齡大���、基礎(chǔ)疾病患者等均可能造成不同程度的不良事件�����,而大容量注射劑由于給藥體積較大且可能存在多日連續(xù)給藥��,因此受可見異物污染引發(fā)的臨床風(fēng)險明顯高于小容量注射劑����。
如果風(fēng)險評估結(jié)果顯示可見異物是由于處方或工藝設(shè)計不佳造成�,申請人應(yīng)主動應(yīng)對風(fēng)險,及早發(fā)現(xiàn)問題��,優(yōu)化產(chǎn)品處方工藝,而不是通過調(diào)整中下游工藝或嚴格終產(chǎn)品放行來達到控制產(chǎn)品的目的���。 在產(chǎn)品開發(fā)階段對可見異物進行風(fēng)險評估���,一方面有助于理解處方工藝,為后續(xù)的持續(xù)改進奠定基礎(chǔ)�;另一方面��,風(fēng)險評估的結(jié)果還可以為確定生產(chǎn)過程中可見異物檢測的警戒限和行動限提供依據(jù)�����,同時將識別出并經(jīng)表征(包括尺寸�����、形狀�����、顏色等) 的可見異物作為陽性樣品用于培訓(xùn)和檢查對照���。
三�����、可見異物檢查
制定全面的檢查程序是注射劑中可見異物控制的一個重要環(huán)節(jié)�����。 注射劑中可見異物檢查一般包括生產(chǎn)過程中 100% 檢查與每批產(chǎn)品基于統(tǒng)計學(xué)抽樣放行檢查�����。 應(yīng)結(jié)合產(chǎn)品特性與臨床風(fēng)險等盡早制定可見異物檢查方案����,包括檢查方法、統(tǒng)計學(xué)抽樣計劃�����、接收/ 拒收標準��、檢查結(jié)果評估標準���、人員培訓(xùn)��、設(shè)備確認與偏差調(diào)查等�����。 不同產(chǎn)品以及制劑處方工藝��、原輔包���、批量等發(fā)生變更等時���,需評估可見異物檢查方案的適用性,隨產(chǎn)品生命周期中的更新隨時調(diào)整����。 通過可見異物檢查應(yīng)確保注射劑中僅存在低檢測概率的異物����。
3.1 生產(chǎn)過程中100%檢查
生產(chǎn)過程中應(yīng)采用經(jīng)過驗證的方法對注射劑中可見異物進行 100% 檢查并剔除不合格品,并應(yīng)在最易檢測到可見異物的工序進行檢查�,例如:一般應(yīng)在貼標簽前進行可見異物檢查,以避免標簽的影響���。
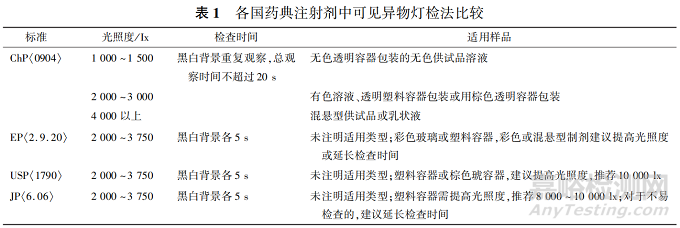
當前���,常用的可見異物檢查方法包括人工燈檢��、半自動化檢查與自動化檢查�;ChP 中僅收載了燈檢法與光散射法[1]�����,其中光散射法屬于自動化檢查范疇�����;EP[20]�����,JP[14]均僅收載了燈檢法�。
3.1.1人工燈檢 人工燈檢是通過人眼視力直接分辨異物,是目前可見異物檢查最常見也是最主要的方法���,各國藥典均有收載��。 人工燈檢的關(guān)鍵參數(shù)主要包括[16]:① 背景和對比�����,采用黑色或白色的純色背景板提供對比��,以最大化產(chǎn)品內(nèi)容物的可見性����。② 光照度與檢查時間,需結(jié)合包裝容器顏色與產(chǎn)品特性等選擇合適的光照度�����,并保證每個容器都有足夠的檢查時間���;如表 1 所示�����,對于一般透明容器,EP�,USP,JP 中規(guī)定的光照度與檢查時間等均無明顯差異�����,ChP 中推薦光照度略低�����,但檢查時間相應(yīng)延長;對于棕色或塑料容器�、混懸型注射劑等,各國藥典均建議提高光照度���,EP 與 JP 還建議可以延長檢查時間��。 ③ 需仔細旋轉(zhuǎn)或倒置內(nèi)容物����,確保容器和瓶塞內(nèi)表面的可見異物得到充分檢查���,溶液型注射劑還應(yīng)盡量避免引入氣泡����,以減少檢測誤差��。
人工燈檢受人為因素影響較大����,靈敏度相對較低,良好的培訓(xùn)和訓(xùn)練可以在一定限度上降低檢查人員的變異性����。 ChP 中明確提出�����,深色透明容器包裝或液體色澤較深(一般深于各標準比色液 7 號)的品種不適合采用人工燈檢法進行可見異物檢查����,應(yīng)采用光散射法等自動化檢查技術(shù)[1]����。
3.1.2 半自動化檢查技術(shù) 將自動化容器處理技術(shù)與人工燈檢結(jié)合,在機械支持下���,容器可以在檢查者面前以恒定速度上下翻轉(zhuǎn)或旋轉(zhuǎn)����,停止后檢查者觀察容器中的可見異物�,機械或手動剔除不合格品[21]。 半自動化檢查技術(shù)與人工燈檢工作原理相同��,關(guān)鍵參數(shù)相似����,但檢查速度需經(jīng)過確認,工作效率明顯提高���。
3.1.3 自動化檢查技術(shù) 采用與半自動化檢測技術(shù)相似的自動化容器處理技術(shù)�����,檢測技術(shù)由“人工檢查”發(fā)展為“自動化檢查”�����。 常見的自動化檢查技術(shù)主要包括高速工業(yè)相機�、可見光二極管陣列��、X?射線����、近場雷達、紫外和近紅外光譜等�����,通過不同波長和傳感器檢測可見異物����,并自動剔除��。 ChP 中的光散射法(第二法)即利用注射劑中的可見異物使入射光發(fā)生散射的原理�����,通過檢測光散射能量����,達到檢查注射劑中可見異物的目的[1]��。
與人工燈檢和半自動化檢查技術(shù)相比���,自動化檢查技術(shù)不僅提高了工作效率����,降低了檢查變異性����,還提高了靈敏度和可靠性,甚至可以檢測到比目檢閾值下限更小的可見異物�����。 對許多無法采用人工燈檢法檢查可見異物的產(chǎn)品可以采用自動化檢查技術(shù),如無菌粉末��、混懸型注射劑或非透明玻璃包裝注射劑等����。 如果可見異物檢查方法從前 2 種方法變更為自動化檢查技術(shù)����,由于靈敏度提高,建議基于數(shù)據(jù)積累與統(tǒng)計學(xué)研究調(diào)整可見異物行動限和警戒限����。自動化檢查技術(shù)的缺點是設(shè)備昂貴、驗證復(fù)雜�����、高靈敏度同時也可能會造成高誤檢率����。
無論選擇何種方法檢查可見異物,都應(yīng)結(jié)合產(chǎn)品特性對檢查參數(shù)進行研究�,如光照度、每個單元檢查時間�����、檢查人員疲勞時間等,根據(jù)研究結(jié)果與檢查情況制定操作規(guī)程����。 完整的可見異物控制和監(jiān)測程序,除包括可見異物檢查操作規(guī)程外��,還應(yīng)包括生產(chǎn)過程控制�����、中間產(chǎn)品檢查����、微生物污染等操作規(guī)程,以最大限度減少可見異物的產(chǎn)生����、識別受影響批次并協(xié)助調(diào)查可見異物來源。
3.1.4 特殊注射劑的考量[16- 17] 由于制劑特性和/ 或包裝容器等特殊性�,部分產(chǎn)品無法采用目檢法檢查可見異物或方法靈敏度降低。 例如:無菌粉針劑中固體粉末內(nèi)容物可能掩蓋可見異物��、混懸型注射劑無法檢出所有可見異物����、大容量注射劑的包裝標簽以及棕色或塑料包裝等均可能干擾目檢�。 對于此類特殊注射劑通常建議增加光照度���、延長檢查時間、放大可見異物或采用靈敏度更高的檢查方法等�,使可見異物得到充分檢查,USP 與 JP 推薦光照度最高達 10 000 lx���。 部分產(chǎn)品由于對光較敏感而無法采用燈檢法�����,例如:對于甲鈷胺注射液�����,即使經(jīng)過燈檢短時間光照�,光降解雜質(zhì)也呈增加趨勢�,一般推薦采用靈敏度更高的自動化檢查技術(shù)。
除上述措施外�����,還可以在 100% 檢查后基于統(tǒng)計學(xué)抽樣增加破壞性補充檢查,為特殊注射劑的可見異物控制提供額外保障���。 例如:將混懸型注射劑過濾除去固有異物后再檢查��、無菌粉針劑復(fù)溶后再檢查���、棕色或不透明容器包裝的產(chǎn)品轉(zhuǎn)移至透明容器中再檢查等。 當然�����,如果能夠證明 100% 檢查所用的可見異物方法能夠達到人工目檢的靈敏度����,則不需要增加破壞性補充檢查。
3.2 基于統(tǒng)計學(xué)抽樣放行檢查
FDA 指南草案明確在生產(chǎn)過程中對注射劑可見異物進行 100% 檢查后�����,還應(yīng)基于統(tǒng)計學(xué)抽樣計劃��,采用經(jīng)過驗證的檢查方法及恰當?shù)目山邮軜藴蔬M行產(chǎn)品放行檢驗��。 推薦參考 ASTM E2234. 22 等擬定抽樣計劃與可接受標準��,同時還需考慮產(chǎn)品特性、批量大小���、患者風(fēng)險�����、可見異物類型與檢測干擾因素(如難以進行可見異物檢查的產(chǎn)品)等��,對于風(fēng)險較高的產(chǎn)品或觀察到非典型缺陷的產(chǎn)品應(yīng)制定更加嚴格的取樣計劃和可接受標準。 如果采用留樣樣品評估市售產(chǎn)品(如產(chǎn)品由于可見異物投訴)����,則應(yīng)將設(shè)施設(shè)備和/ 或該產(chǎn)品的歷史檢查數(shù)據(jù)等一并考慮在內(nèi)。 如果單個容器內(nèi)存在多個可見異物��,表明可能存在生產(chǎn)問題�����,應(yīng)對該批次進行更加嚴格的可見異物檢查����。
相比之下,ChP 則明確規(guī)定了可見異物檢查的取樣數(shù)量�,注射液一般取 20 支(瓶)�,注射用無菌制劑一般取 5 支(瓶)�,并制定了明顯可見異物、微細可見異物數(shù)量檢出的可接受標準[1]��。這與FDA 指南草案[11]�����、USP[12] 的相關(guān)規(guī)定存在較大差異��,ChP按照固定樣品量進行抽樣檢查����,未將產(chǎn)品批量、患者臨床風(fēng)險�����、產(chǎn)品特性等因素考慮在內(nèi)�����,不僅給藥品國際化生產(chǎn)和監(jiān)管帶來了極大的挑戰(zhàn)�,也難以有效、科學(xué)地控制可見異物��。
3.3 培訓(xùn)和確認
人員培訓(xùn)與設(shè)備確認等對確保可見異物檢查的穩(wěn)健性很重要�����。 可見異物檢查人員需定期培訓(xùn)考核�����,用于可見異物檢查的設(shè)備需定期驗證確認��,以最大限度減少不同人員或設(shè)備造成的可見異物檢查結(jié)果差異����。 建議將工藝開發(fā)中識別出的可見異物����、生產(chǎn)過程中或放行檢驗時檢測到以及制備或購買的可見異物缺陷樣品收集起來,與合格樣品混合后用于人員培訓(xùn)或設(shè)備確認���,培訓(xùn)樣品總量需滿足統(tǒng)計學(xué)要求�����,可見異物缺陷樣品不應(yīng)超過總量的10% ~20% ��,可見異物缺陷樣品應(yīng)具有代表性���,且應(yīng)包含目檢閾值下限的缺陷樣品����,以確認檢查靈敏度�����。 隨著生產(chǎn)檢驗過程中逐漸識別出新可見異物�,可不斷擴充培訓(xùn)和檢查對照樣品。
3.4 偏差調(diào)查
應(yīng)對生產(chǎn)過程中或放行檢驗中檢出的可見異物不合格品�����、上市后投訴召回的可見異物不合格品等引發(fā)的質(zhì)量偏差進行調(diào)查�,必要時將調(diào)查范圍擴展到其他批次。 偏差調(diào)查一般包括加強取樣計劃�����、調(diào)查可見異物潛在來源并評估對產(chǎn)品放行的影響等����。通過偏差調(diào)查應(yīng)盡量識別出可見異物并分類���,確認發(fā)生原因的同時提出有針對性的糾正和預(yù)防措施,如優(yōu)化處方工藝�����、改進可見異物檢查程序等��。 偏差調(diào)查是可見異物產(chǎn)品生命周期管理中的重要環(huán)節(jié)�,在可見異物控制策略中發(fā)揮著重要作用。
四����、基于產(chǎn)品生命周期方法的可見異物控制
可見異物控制開始于產(chǎn)品開發(fā)階段,應(yīng)持續(xù)貫穿整個產(chǎn)品生命周期��,包括貯藏期間穩(wěn)定性評價與上市后產(chǎn)品監(jiān)控��。 具體研究內(nèi)容涉及原輔包選擇和質(zhì)量控制��、設(shè)施設(shè)備選擇��、產(chǎn)品開發(fā)���、產(chǎn)品生產(chǎn)過程控制(如中間過程取樣控制���、100% 檢查等)、產(chǎn)品放行檢驗���、貯藏期間及臨床使用中可見異物考察等�����,是一個不斷持續(xù)的質(zhì)量確認和改進過程��。 生產(chǎn)過程中的相關(guān)信息����,如生產(chǎn)偏差����、檢查缺陷、設(shè)備設(shè)施故障等����,可以提供生產(chǎn)過程中可見異物控制的狀態(tài)信息、產(chǎn)品質(zhì)量指標(如穩(wěn)定性考察結(jié)果��、產(chǎn)品投訴召回等)及產(chǎn)品不良反應(yīng)報告可以提示與可見異物相關(guān)的產(chǎn)品質(zhì)量問題?���?傊a(chǎn)過程中����、貯藏及上市后的產(chǎn)品反饋信息,如可見異物趨勢增加�����、出現(xiàn)新可見異物��、超過警戒限或行動限等����,一方面可以評估可見異物控制策略的有效性,另一方面也可以提示產(chǎn)品處方工藝設(shè)計是否存在缺陷��。 如果調(diào)查發(fā)現(xiàn)產(chǎn)品處方或工藝設(shè)計存在缺陷���,應(yīng)重新進行設(shè)計以確保產(chǎn)品質(zhì)量可控�����。
五��、結(jié)語
根據(jù)美國 FDA 指南的要求����,可見異物從來源和性質(zhì)分為固有異物�、內(nèi)源性異物和外源性異物,其產(chǎn)生與產(chǎn)品處方工藝�����、原輔包����、生產(chǎn)設(shè)備等密切相關(guān),還與注射劑的生產(chǎn)工藝控制和 GMP 實施息息相關(guān)����。國內(nèi)外均規(guī)定注射劑應(yīng)在符合 GMP 的條件下生產(chǎn),并在生產(chǎn)過程中進行 100% 檢查以確?���?梢姰愇锓弦蟆?但由于可見異物來源和組成復(fù)雜����,且大多具有概率性���,加上目視檢查方法存在的缺陷,導(dǎo)致生產(chǎn)過程中可見異物不合格率居高不下并經(jīng)常出現(xiàn)放行后的注射劑可見異物檢查不合格�����。
目前�,國內(nèi)業(yè)界對注射劑中可見異物的控制未引起足夠重視,過度依賴成品檢驗����,忽視了對可見異物的風(fēng)險評估和全生命周期的管理,很少結(jié)合產(chǎn)品特性����、可見異物特征及臨床風(fēng)險等制定有針對性的控制策略。 目前在參照 ChP 標準進行生產(chǎn)和審評的基礎(chǔ)上�,僅成品檢驗結(jié)果符合藥典標準規(guī)定已難以滿足注射劑中可見異物的控制要求。 建議結(jié)合FDA 指南草案并遵循 ICH Q8�����,Q9��,Q12 等研究理念,在嚴格按照 GMP 要求進行注射劑生產(chǎn)的基礎(chǔ)上�����,從源頭把關(guān)����,基于風(fēng)險識別和評估��,針對不同產(chǎn)品���、不同類型可見異物建立相應(yīng)的控制策略����,重視過程控制�����,基于風(fēng)險評估采用統(tǒng)計學(xué)抽樣檢驗進行產(chǎn)品放行��,根據(jù)后續(xù)反饋信息����,對處方工藝與可見異物控制策略等持續(xù)改進完善��,建立整體的��、全生命周期管理的可見異物控制策略���,確保注射劑中可見異物可控。