摘要
目的:對2020年新修訂的《藥品注冊管理辦法》頒布前后我國納入藥品加快上市注冊程序并已批準(zhǔn)上市的藥品信息進行分析,為完善我國藥品加快上市注冊程序提供參考����。
方法:檢索國家藥品監(jiān)督管理局藥品審評中心發(fā)布的2019-2021年度藥品審評報告,對通過優(yōu)先審評和藥品加快上市注冊程序而上市的相關(guān)藥品數(shù)據(jù)資料進行信息整理和匯總分析��。
結(jié)果:通過藥品加快上市注冊程序的上市藥品數(shù)量逐年增多��,優(yōu)先審評審批資源向臨床優(yōu)勢藥品和創(chuàng)新藥品傾斜����。
結(jié)論:藥品加快上市注冊程序可為臨床價值顯著的藥品提高審批效率,在一定程度上激發(fā)制藥企業(yè)研制熱情����,但藥品加快上市注冊審批體系仍需進一步完善配套政策����,細化實施要求����,加快專業(yè)指南出臺鼓勵以臨床價值為導(dǎo)向的藥品的研發(fā)創(chuàng)制,不斷提升藥品監(jiān)管部門的服務(wù)和監(jiān)管能力��。
隨著我國藥品審評審批制度改革的不斷推進��,對多元化的激勵藥品研發(fā)創(chuàng)制途徑的需要日益迫切��。2019年新修訂的《藥品管理法》中明確指出“支持以臨床價值為導(dǎo)向的藥物創(chuàng)新��,推動藥品技術(shù)進步”���,2020年新修訂的《藥品注冊管理辦法》(以下簡稱《辦法》)進一步提出“要建立并實施加快藥品上市注冊程序。”在新的法規(guī)框架下�,加快上市注冊申請覆蓋了臨床試驗和上市許可的全過程,而更加細化的程序和要求有便于行業(yè)主體執(zhí)行����,推動藥品創(chuàng)新,加快以臨床價值為導(dǎo)向的藥品的審評審批速度�。本文以《辦法》中藥品加快上市注冊程序為切入點�����,分析通過藥品加快上市注冊程序的上市藥品數(shù)據(jù)信息����,總結(jié)當(dāng)前階段我國藥品加快審評審批政策落地實施情況���,為完善我國藥品加快上市注冊程序以及推進以臨床價值為導(dǎo)向的藥品的創(chuàng)新提供建議和參考����。
一�����、數(shù)據(jù)來源
通過查詢國家藥品監(jiān)督管理局藥品審評中心(Centerfor Drug Evaluation��,CDE)網(wǎng)站2019-2021年的藥品審評報告和信息公開欄�����,整理研究所用的藥品申請��、納入和通過程序的件數(shù)(以注冊申請件數(shù)計)以及藥品品種個數(shù)(以通用名稱計)等信息。主要根據(jù)2019年通過優(yōu)先審評上市的藥品數(shù)據(jù)�����,2020年納入加快上市程序并已批準(zhǔn)上市藥品的數(shù)據(jù)以及2021年部分公開的通過程序并已批準(zhǔn)上市的藥品數(shù)據(jù)����,并按照藥品加快上市注冊程序,對申請�、納入并通過上市的申請數(shù)量、藥品類型以及重點治療領(lǐng)域等方面進行數(shù)據(jù)處理和統(tǒng)計分析���。
二����、結(jié)果與分析
2.1 藥品加快上市注冊的程序路徑分布總體情況
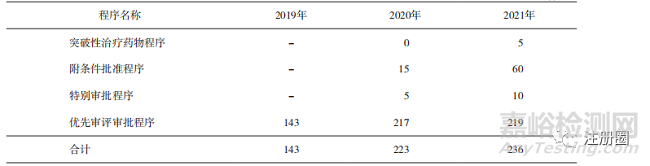
2019-2021年通過加快上市程序批準(zhǔn)上市的申請數(shù)量持續(xù)增加��,其中優(yōu)先審評審批程序申請數(shù)量最多��,突破性治療藥物程序申請數(shù)量最少(詳見表1)?��,F(xiàn)有制度構(gòu)建的加快上市途徑對于促進臨床價值突出和公共衛(wèi)生急需等藥品的上市具有明顯推動作用。
▲ 表1-已批準(zhǔn)上市納入藥品加快上市注冊程序的數(shù)量統(tǒng)計 件
注:存在一個注冊申請同時申請多個程序情況���,合計值并非求和項���。
2.2 突破性治療藥物程序申請注冊情況
突破性治療藥物程序適用于通過早期臨床研究數(shù)據(jù)發(fā)現(xiàn)具有突出臨床優(yōu)勢��,可以此縮短臨床研發(fā)周期的藥物��。與國際上類似的藥品加快上市注冊程序相比���,我國突破性治療藥物程序施行時間尚短,還處在探索階段��。2012年美國食品藥品管理局(Food and Drug Administration�,F(xiàn)DA)開始實施突破性治療認定(Breakthrough Therapy Designation����,BTD),歐洲藥品管理局(European MedicinesAgency�����,EMA)在2016年實行了優(yōu)先藥物計劃(Priority Medicines��,PRIME)。
2.2.1 突破性治療藥物程序關(guān)注度較高
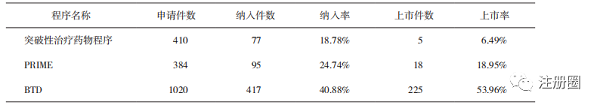
截至2021年12月31日,突破性治療程序累計申請410件��,CDE納入77件突破性治療藥物申請����,其中5件已批準(zhǔn)上市����。
對比截至2021年底FDA公布的BTD相關(guān)數(shù)據(jù)[1]以及EMA公布的PRIME相關(guān)數(shù)據(jù)[2]����,我國僅實施2年的突破性治療藥物程序申請總量比實施6年的PRIME申請總量還要多26件�����,是施行10年的BTD申請總量的40.20%(詳見表2)�,證明《辦法》實施后的政策獲益對于我國制藥企業(yè)具有較強的吸引和導(dǎo)向作用�。
從審評時限看��,與正常序列品種200日的常規(guī)上市許可申請的審評時限相比�����,從臨床試驗階段申請納入突破性治療藥物程序到批準(zhǔn)上市僅在241~371日之間��,可見突破性治療藥物從研發(fā)到上市的進程明顯加快。
▲ 表2-我國突破性治療藥物程序���、PRIME 和 BTD 的數(shù)量統(tǒng)計
2.2.2 突破性治療藥物程序的納入率和上市批準(zhǔn)率相對較低
我國突破性治療程序的納入率和上市批準(zhǔn)率分別為18.78%和6.49%,明顯低于美國和歐盟����,說明相關(guān)申請的針對性較低�����,與政策的契合度有待提高���。這與政策實施時間較短,政策的細化內(nèi)容尚不深入有關(guān)�,例如EMA細化了不同的研發(fā)階段����,包括早期臨床和臨床研發(fā)階段的技術(shù)要求,我國僅明確了需提交的資料內(nèi)容�����,對于企業(yè)早期臨床研發(fā)缺乏指導(dǎo)作用����;FDA則要求審評人員主動發(fā)現(xiàn)滿足BTD的藥物�����,同時強調(diào)有經(jīng)驗的審評人員參會以及除正式會議外要保持意見交換���,推動BTD認證和加快上市���,目前我國還沒有類似的措施出臺[3]�。
2.3 附條件批準(zhǔn)程序申請注冊情況
附條件批準(zhǔn)程序適用于尚未完成完整臨床研究時����,通過“先批準(zhǔn)后驗證”的形式加快具有突出臨床價值的臨床急需藥品上市[4],旨在縮短藥物臨床研發(fā)時間�,附“條件”盡快將優(yōu)勢產(chǎn)品推向市場。
2.3.1 附條件批準(zhǔn)程序獲益初顯
截至2021年12月31日��,已有75件注冊申請附條件批準(zhǔn)上市��。與新藥上市申請(New DrugApplication,NDA)數(shù)據(jù)對比來看����,2020-2021年上市的NDA申請增長率為55.29%��,附條件批準(zhǔn)上市申請件數(shù)增長率為300%����。經(jīng)附條件批準(zhǔn)的上市申請占NDA的比例也由2020年的7.21%增加至2021年的18.58%�,增長速度明顯加快且占比越來越高����。
與同為新增快速上市途徑的突破性治療藥物程序相比���,附條件批準(zhǔn)的上市申請數(shù)量明顯較多���,這主要因為附條件批準(zhǔn)基于替代終點�����、中間臨床終點或早期臨床試驗數(shù)據(jù)批準(zhǔn)上市����,上市周期更短��,引發(fā)了行業(yè)的更大關(guān)注�。
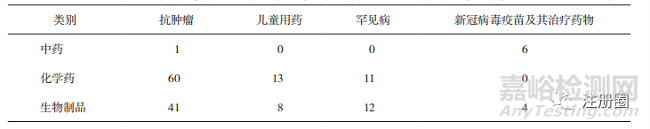
從重點治療領(lǐng)域看����,腫瘤治療申請58件�,占附條件批準(zhǔn)程序申請的77.33%,新冠病毒疫苗申請8件�,占已上市新冠病毒疫苗及其治療藥物(15件)的53.33%��,附條件批準(zhǔn)符合程序設(shè)計定位,有助于將醫(yī)療急需�、臨床價值顯著的藥品提前推向市場�����,獲益初顯�。
2.3.2 附條件批準(zhǔn)程序存在用藥安全潛在風(fēng)險
我國制度實施時間尚短�����,符合附條件批準(zhǔn)程序的藥物研發(fā)與技術(shù)審評都處于探索階段��。在已上市的75件附條件批準(zhǔn)申請中�,僅有5個品種轉(zhuǎn)為正式批準(zhǔn)[5]����,絕大多數(shù)申請還在上市后研究進程中。
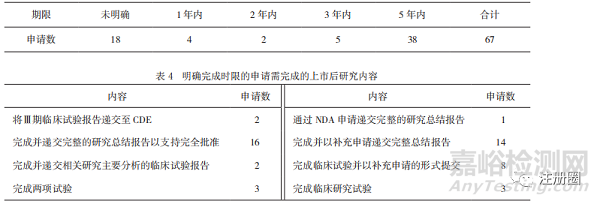
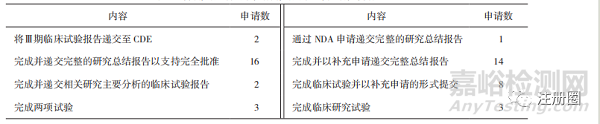
分析除新冠病毒疫苗和治療藥物外已公開的67份申請上市技術(shù)審評報告�����,其中在49件報告中�,CDE明確了完成后續(xù)研究的期限和相關(guān)研究內(nèi)(詳見表3、表4)�。未明確時限的上市申請僅寫明了需要上市許可人完成的研究內(nèi)容,少部分規(guī)定了與CDE溝通臨床試驗進展的時間�����,作為附“條件”上市的藥品�����,上市后監(jiān)管時限的缺失可能會導(dǎo)致公眾臨床用藥安全問題出現(xiàn)��。
▲ 表3-上市后要求完成期限
▲ 表4-明確完成時限的申請需完成的上市后研究內(nèi)容
2.4 特別審批程序申請注冊情況
特別審批程序是國家藥品監(jiān)督管理局依法將臨床急需的保障公眾生命健康的藥品加速推向市場的程序設(shè)計�,主要用于應(yīng)對突發(fā)公共衛(wèi)生事件�����。
2.4.1 特別審批程序主要集中于應(yīng)急藥品需求
截至2021年12月31日�,共納入特別審批程序140件��,均為新冠病毒疫苗及其治療藥物�,批準(zhǔn)臨床試驗申請80件,批準(zhǔn)上市申請15件��,批準(zhǔn)補充申請44件��。
2.4.2 特別審評程序上市速度明顯加快
相較于新藥研發(fā)到上市一般需要10年以上的耗時���,而我國獲批上市的4款新冠病毒疫苗����,在以統(tǒng)一指揮和早期介入為原則的特別審批程序運行下��,僅用不到2年時間可實現(xiàn)從研發(fā)到上市���。
2.5 優(yōu)先審評審批程序申請注冊情況
為解決藥品注冊申請積壓問題���,2015年國家啟動藥品審評審批改革����,先后頒布了《總局關(guān)于解決藥品注冊申請積壓實行優(yōu)先審評審批的意見》和《總局關(guān)于鼓勵藥品創(chuàng)新實行優(yōu)先審評審批的意見》��,提出了優(yōu)先審評并明確申請范圍����。2020年《辦法》出臺后�����,優(yōu)先審評審批程序的申請范圍和條件較前期政策有了明顯變化�,不再以治療具體疾病納入���,剔除占用大量優(yōu)先審評資源卻未體現(xiàn)出臨床價值和市場短缺的仿制藥[6],優(yōu)先審評資源更加凸顯臨床優(yōu)勢�。
2.5.1 優(yōu)先審評審批程序作用明顯
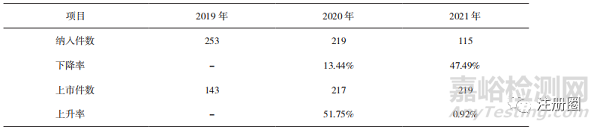
納入優(yōu)先審評審批程序的申請數(shù)量逐年減少且降幅明顯加快(詳見表5)�,獲批上市的申請數(shù)量則逐年增加��,表明監(jiān)管部門優(yōu)先審評資源投入更集中��,提升了優(yōu)先審評審批的效率���,保障了臨床優(yōu)勢藥品和創(chuàng)新藥品的盡快上市��。
▲ 表5-2019-2021 年納入和通過優(yōu)先審評審批程序的申請數(shù)量及變化情況
2.5.2 納入優(yōu)先審評審批程序各類情形
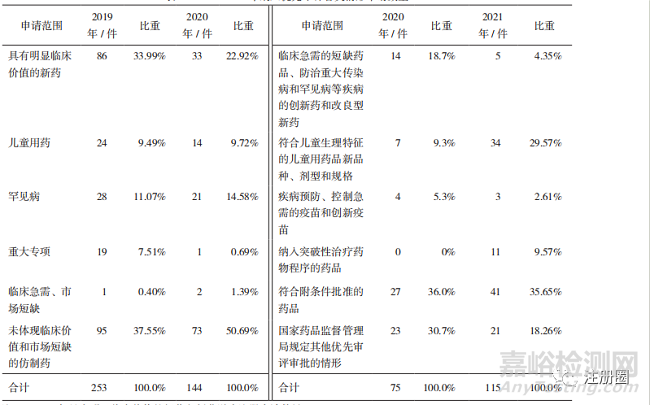
《辦法》的實施����,優(yōu)先審評審批程序呈現(xiàn)縮口狀態(tài)�,政策紅利向臨床急需的用藥傾斜,例如附條件批準(zhǔn)程序藥品���、兒童用藥�、罕見病用藥等�,詳見表6��。
▲ 表6-2019-2021 年納入優(yōu)先審評各類情形申請數(shù)量
注:1. 2020 年具有明顯臨床價值的新藥包括艾滋病注冊申請數(shù)量����。
2. 未體現(xiàn)臨床價值和市場短缺的仿制藥包括同步申報、按與原研藥質(zhì)量和療效一致的標(biāo)準(zhǔn)完善后重新申報��、專利到期和首仿藥品�。
2.6 藥品加快上市注冊程序的藥品類型分布
根據(jù)《辦法》對于藥品注冊管理的分類�,將藥品分為中藥、化學(xué)藥品和生物制品3個類型進行統(tǒng)計分析��。
2.6.1 各類型藥品加快上市注冊的總體情況
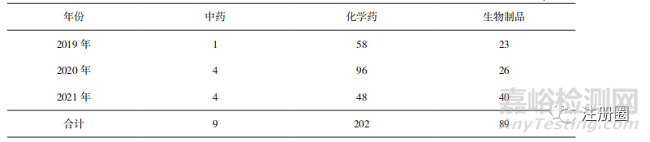
通過藥品加速上市注冊程序的化學(xué)藥品數(shù)量最多��,生物制品次之���,中藥品種數(shù)量最少(詳見表7)���,這和常規(guī)審評上市注冊情況一致����。生物制品作為未來制藥行業(yè)的創(chuàng)新熱點領(lǐng)域���,納入并通過藥品加快上市注冊程序的數(shù)量不斷增加��。
▲ 表7-2019-2021 年通過藥品加快上市注冊程序不同藥品類別上市品種數(shù)量 個
2.6.2 各類型藥品加快上市注冊的比較分析
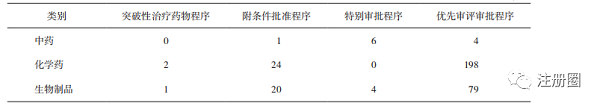
中藥品種以特別審批程序為主(詳見表8),以清肺排毒顆粒��、化濕敗毒顆粒和宣肺敗毒顆粒為經(jīng)典名方代表的審評實踐����,加快了中藥上市的進程�����,“三方”的獲批上市是中藥監(jiān)管科學(xué)發(fā)展的一個標(biāo)志性成果[7]�����,也充分發(fā)揮了中藥品種對于新冠病毒的治療優(yōu)勢����。除新冠治療藥物外,僅有1個腫瘤治療藥物����,兒童用藥等中藥傳統(tǒng)優(yōu)勢病種的相關(guān)申請相對不足(詳見表9)��。
▲ 表8-中藥�����、化學(xué)藥及生物制品通過四種途徑的上市品種數(shù)量 個
▲ 表9-不同藥品類別通過藥品加快上市注冊程序在重點治療領(lǐng)域的上市品種數(shù)量 個
化學(xué)藥品和生物制品以優(yōu)先審評審批程序為主��,這是因為與作為鼓勵真正的創(chuàng)新藥投入臨床使用的突破性治療藥物程序和附條件批準(zhǔn)程序相比��,優(yōu)先審評審批程序政策納入口徑相對寬泛,申請難度相對較小���。在重點治療領(lǐng)域����,二者在兒童用藥���、罕見病治療等方面數(shù)量相當(dāng)��,而在腫瘤治療方面���,盡管化學(xué)藥品在數(shù)量上比生物制品多,但增長速度上生物制品超過了化學(xué)藥品����,這與生物制品相關(guān)的生物治療是繼手術(shù)����、放療和化療之后的第四代腫瘤治療方法[8]相關(guān)����,但有研究表明����,境內(nèi)生產(chǎn)的生物制品中自主研發(fā)的1類創(chuàng)新藥比重低��,且像PD-1單抗的研發(fā)扎堆明顯,與國際先進水平還有一定差距[9]��。
三��、討論
藥品加快上市注冊程序彌補了我國從藥物研發(fā)到上市監(jiān)管的全生命周期覆蓋制度設(shè)計的缺失��,以臨床價值為導(dǎo)向注重藥物創(chuàng)新,縮短藥物研發(fā)時間��,提高審評效率���,政策實施取得了階段性效果����。一方面����,優(yōu)先配置審評資源,與藥品監(jiān)管部門進行溝通以及縮短審評時限加速上市等政策獲益�,激發(fā)企業(yè)研發(fā)熱情���,提高創(chuàng)制質(zhì)量���;另一方面��,藥品可以更快投入臨床應(yīng)用�,增加治療選擇��,保障公眾用藥可及性�。
但由于《辦法》出臺時間短�,新舊政策交替過渡���,藥品加快上市注冊程序?qū)嵤┻€在探索階段����。幫助企業(yè)突破技術(shù)壁壘���、加快研發(fā)成果轉(zhuǎn)化���、加快審評制度改革完善、提供切實對企業(yè)創(chuàng)新形成激勵作用的政策紅利等需要藥品監(jiān)管部門重點關(guān)注����。
3.1 藥品加快上市注冊程序相關(guān)制度有待進一步完善
從出臺的藥品加快上市注冊程序配套文件來看,缺乏原則性政策的具體操作文件�,沒有形成通用性指南,不能很好地指導(dǎo)企業(yè)執(zhí)行政策�����,甚至?xí)?dǎo)致審評效率低下���。在《辦法》中僅涉及了特別審批程序的原則性表述����,并未更新工作程序和技術(shù)指導(dǎo)���,附條件批準(zhǔn)程序也尚未出臺上市后監(jiān)管具體要求。對比國際先進做法�����,審評人員在臨床急需用藥上市注冊中主導(dǎo)作用發(fā)揮不充分��,審評資源利用不充分、不高效����。
3.2 藥品創(chuàng)新激勵有待進一步加強
我國原創(chuàng)性新藥數(shù)量相對少,國產(chǎn)創(chuàng)新藥處在重要轉(zhuǎn)型期����,國內(nèi)藥品領(lǐng)域存在過于激烈的同質(zhì)化競爭����,上市產(chǎn)品差異化不明顯[10]。藥品加快上市注冊程序作為我國藥品加速審批路徑�,注重納入和上市注冊藥品的臨床價值和創(chuàng)新性����,但目前來看,藥品臨床優(yōu)勢療效較藥品的創(chuàng)新性在4種加速途徑中占比更多�。
四�、建議
4.1 持續(xù)完善藥品加快上市注冊程序制度改革
4.1.1 加快專業(yè)技術(shù)指南出臺
現(xiàn)有加快上市注冊制度的構(gòu)建是為了解決臨床急需用藥問題。對比常規(guī)審批上市注冊藥物����,通過藥品加快上市注冊程序上市的藥品臨床研發(fā)難度大,臨床設(shè)計方案難點多��。建議盡快出臺能夠作為通用規(guī)范的技術(shù)指南��,如藥學(xué)通用技術(shù)指南,提供充足的技術(shù)保障���,以服務(wù)于藥學(xué)研究與技術(shù)審評[11]??偨Y(jié)審評經(jīng)驗和不同適應(yīng)證特點�,考慮藥物本身創(chuàng)新程度及作用機制,規(guī)范臨床試驗設(shè)計方案���,內(nèi)容方面可以涉及用于不同研究的合理樣本量,在無法滿足樣本數(shù)時建議采用的研究方法及資料完整度要求等����,減少企業(yè)因臨床設(shè)計方案缺陷導(dǎo)致納入程序批準(zhǔn)上市失敗。
4.1.2 推進相關(guān)配套政策完善
政策文件需要貼合當(dāng)前環(huán)境發(fā)展以及事物本身發(fā)展規(guī)律。建議藥品監(jiān)管部門盡快更新特別審批程序的工作程序�,補充申報資料要求,明確溝通機制�,建立退出和終止程序,設(shè)置滾動提交審評資料以充分體現(xiàn)審評前置作用���,授予應(yīng)急情況下的賠償責(zé)任豁免權(quán),加快應(yīng)對重大公共衛(wèi)生事件的藥品上市�,使得特別審批更具可操作性��;出臺附條件批準(zhǔn)上市后監(jiān)管法規(guī)文件�,針對藥品研發(fā)特點���,合理明確上市后確證性研究期限時長和規(guī)范化審評標(biāo)準(zhǔn)����,以完整上市后審評路徑�;不斷開發(fā)符合中藥特點的審批制度����,如《中藥注冊管理專門規(guī)定》的出臺可以幫助企業(yè)厘清中藥研發(fā)的思路��,從制度設(shè)計上提高中藥審評審批效率。在未來政策完善過程中應(yīng)尊重中藥品種發(fā)展規(guī)律�,建議可依據(jù)實際情況來看待藥效學(xué)研究����。同時�,中藥新藥研發(fā)要避免只關(guān)注重大疾病,應(yīng)聚焦常見病�、多發(fā)病�����,充分發(fā)揮中藥臨床優(yōu)勢。
4.1.3 細化政策實施原則性要求
建議藥品監(jiān)管部門可借鑒國際加快藥品上市制度的先進做法����,針對不同研發(fā)階段提出不同技術(shù)的具體要求,定期發(fā)布建議的臨床替代終點指標(biāo)用于企業(yè)研發(fā)參考[12]����,加快我國技術(shù)指導(dǎo)體系與國際通行規(guī)則接軌,更好地發(fā)揮突破性治療藥物程序和附條件批準(zhǔn)藥物程序在藥品上市注冊中前端和中端的加速作用,從臨床研發(fā)早期給予優(yōu)先審評資源�,在一定程度上確保企業(yè)提出滿足監(jiān)管和藥品安全有效的臨床研究策略,提高新藥研發(fā)成功率����,盡快批準(zhǔn)臨床優(yōu)勢產(chǎn)品上市。
4.2 加強藥品監(jiān)管部門服務(wù)和能力建設(shè)
在確保藥品安全性和有效性的前提下����,加強審評隊伍建設(shè)和條件建設(shè)����,改善藥品監(jiān)管部門與申請人的互動交流效果��,提升審評審批效率[13]����。建議藥品監(jiān)管部門不斷推進人員隊伍建設(shè)�����,適當(dāng)擴容審評人員數(shù)量���,合理配置人員工作內(nèi)容��,積極跟進臨床急需用藥上市注冊情況�。開展專業(yè)知識培訓(xùn)并予以考核驗收成果���,強化人員隊伍專業(yè)知識能力�、審批工作完成能力以及藥品審評全流程監(jiān)管核查能力���。同時����,更好地引導(dǎo)藥品研發(fā)企業(yè)正確認識制度要求�,把握政策改革內(nèi)涵,激發(fā)市場主體活力���。
加快藥品上市注冊程序的實施效果初顯�����,審評標(biāo)準(zhǔn)也在進一步探索發(fā)展���。建議規(guī)范技術(shù)審評尺度�,形成制度化行業(yè)共識�����,結(jié)合技術(shù)指南建設(shè)����,不斷在實踐中清晰審評要求,提升藥品審評服務(wù)和監(jiān)管能力���,落實政府“放管服”工作原則���。
4.3 激勵企業(yè)加快臨床價值藥品研發(fā)創(chuàng)制
新增加的突破性治療藥物程序和附條件批準(zhǔn)程序,作為鼓勵真正的創(chuàng)新藥品投入臨床使用����,而非“mebetter”“mebest”藥品����。藥品研發(fā)創(chuàng)新不能僅在現(xiàn)有靶點上開發(fā)新的適應(yīng)證,建議加強原創(chuàng)基礎(chǔ)研究,強化數(shù)字醫(yī)療在醫(yī)藥領(lǐng)域研究應(yīng)用���,研制創(chuàng)新所需大型設(shè)備[14]���,構(gòu)建企業(yè)、高校與醫(yī)院等多方知識交流平臺�����,突破研發(fā)技術(shù)壁壘和研制藥品新靶點�。建議完善同情用藥制度,明確主體參與條件責(zé)任并加強藥品風(fēng)險管理細化實施條件�,彌補創(chuàng)新藥研發(fā)設(shè)計上可能存在的現(xiàn)實問題,配合藥品加快上市注冊程序�����,確保研究成果落地�����,產(chǎn)出具有國際影響力的原創(chuàng)性新藥��。
藥品研發(fā)生產(chǎn)企業(yè)是藥品領(lǐng)域重要的參與主體�����,建議藥品監(jiān)管部門積極尋求行業(yè)對話,建立制藥行業(yè)政府機構(gòu)和專家委員會的對話機制��,就行業(yè)技術(shù)指南���、政策實施效果反饋以及亟待解決領(lǐng)域問題等定期進行意見交換�,激發(fā)企業(yè)主動作為����。同時,藥品監(jiān)管部門及時把握行業(yè)動態(tài)�,予以政策扶持,建議同靶點首家國產(chǎn)創(chuàng)新藥可酌情加快審評審批[15]�����。
當(dāng)前腫瘤治療藥品熱度過高��,存在創(chuàng)新藥靶點研究重復(fù)度高的現(xiàn)象���,建議與新藥研發(fā)專利制度以及兒童用藥市場獨占權(quán)等形成政策鏈接����,鼓勵企業(yè)適當(dāng)開發(fā)腫瘤治療藥物以外的其他領(lǐng)域藥品����。
參考文獻
[1] FDA. Breakthrough Therapy Designation Requests[EB/OL].(2023-01-12)[2023-03-16]. https://www.fda.gov/drugs/ind-activity/breakthrough-therapy-designation requests.
[2] EMA. PRIME:Analysis of the First 5 Years’ Experience[EB/OL].(2022-03-03)[2023-03-16].https://www.ema.europa.eu/en/documents/report/prime analysis-first-5-years-experience_en.pdf.
[3] 袁利佳,汪小燕�����,楊志敏����,等. 突破性治療藥物程序在藥品注冊體系中的作用及展望[J]. 中國藥事,2022��,36(9):973-983.
[4] 唐輝����,湯立達. 藥品附條件批準(zhǔn)上市的內(nèi)涵與風(fēng)險防控[J]. 中國醫(yī)藥導(dǎo)刊,2021����,23(4):289-295.
[5] 袁利佳,陳小明�����,張寧. 我國藥品附條件批準(zhǔn)程序?qū)嵤┣闆r及相關(guān)思考[J]. 中國藥事,2022�����,36(10):1093-1102.
[6] 吳忠虹��,董麗. 我國藥品優(yōu)先審評審批制度的現(xiàn)狀分析[J]. 中國處方藥����,2022,20(10):29-31.
[7] 黃明����,楊豐文,張俊華��,等. 新時代中藥傳承創(chuàng)新發(fā)展呼喚科學(xué)監(jiān)管[J]. 中國中藥雜志����,2023,48(1):1-4.
[8] 謝華玲���,陳芳���,Cynthia���,等. 全球生物制藥領(lǐng)域研發(fā)態(tài)勢分析[J]. 中國生物工程雜志,2019�����,39(5):1-10.
[9] 辛中帥�,張輝�,楊建紅,等. 我國已上市治療用生物制品問題分析及監(jiān)管建議[J]. 中國藥事�����,2019��,33(9):982-985.
[10] 杜逸航�,孫友松,陳倩�,等. 2021年全球獲批上市的原創(chuàng)新藥:回顧與展望[J]. 中國新藥雜志,2022��,31(11):1033-1041.
[11] 趙一飛����,耿欣��,徐立華����,等. 對我國附條件批準(zhǔn)上市化藥藥學(xué)技術(shù)要求的思考[J]. 中國藥科大學(xué)學(xué)報���,2021��,52(5):636-642.
[12] FDA. Table of Surrogate Endpoints That Were the Basis of Drug Approval or Licensure[EB/OL].(2022-02-28)[2023-03-16].
https://www.fda.gov/drugs/development resources/table-surrogate-endpoints-were-basis-drug approval-or-licensure.
[13] 姚立新�,李茂忠��,董江萍����,等. 從PDUFAⅠ到PDUFAV——FDA通過法規(guī)體系的完善實現(xiàn)新藥審評的持續(xù)改進[J]. 中國新藥雜志,2013����,22(10):1143-1156,1169.
[14] 陳麗湘. 加快中醫(yī)藥振興發(fā)展推動中藥新藥盡早上市[N]. 證券時報���,2022-03-10(A04).
[15] 馬立敏��,汪祥波. 深化中藥審評審批改革加快中藥新藥研發(fā)[N]. 南方日報����,2022-03-11(A06).