每個(gè)醫(yī)療器械制造商的目標(biāo)是保持一致的質(zhì)量�,并保證一定水平的安全和有效性。為了實(shí)現(xiàn)這一點(diǎn)���,制造商必須遵守一系列法律���、標(biāo)準(zhǔn)和法規(guī)�����。遵守所有法規(guī)的目的是確?��;颊摺⒂脩艉偷谌降漠a(chǎn)品安全�����。證明技術(shù)文件符合所有必要的要求是產(chǎn)品在特定市場(chǎng)成功注冊(cè)的關(guān)鍵�。
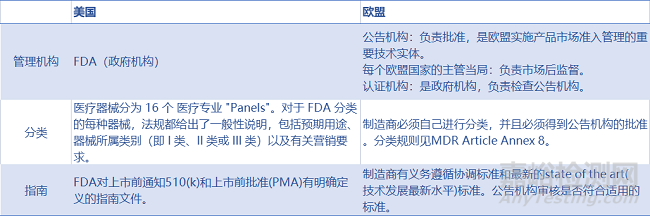
美國(guó)食品和藥物管理局(FDA)是美國(guó)的一個(gè)監(jiān)管機(jī)構(gòu),負(fù)責(zé)批準(zhǔn)即將進(jìn)入美國(guó)市場(chǎng)的醫(yī)療器械���。歐盟醫(yī)療器械法規(guī)是由歐盟委員會(huì)公布的一套法規(guī)���,以指導(dǎo)醫(yī)療器械制造商在將醫(yī)療器械投放市場(chǎng)之前符合法規(guī)要求。FDA法規(guī)和歐盟MDR對(duì)各自的市場(chǎng)都是強(qiáng)制性的�����。
通常情況下,在美國(guó)上市的醫(yī)療器械希望在歐盟市場(chǎng)注冊(cè)并銷售其醫(yī)療器械���,反之亦然���。在這種情況下,最大的問題是需了解兩方市場(chǎng)所要求的技術(shù)文件和上市路徑的異同�����。今天分別介紹FDA和CE法規(guī)要求的異同�,讓您發(fā)現(xiàn)醫(yī)療器械注冊(cè)過程的主要差異�,以及兩類監(jiān)管之間的相似之處。對(duì)于希望在兩個(gè)市場(chǎng)上銷售產(chǎn)品的制造商來說�����,了解哪些文件可以被兩個(gè)市場(chǎng)使用���,以及哪些文件是特定于某個(gè)市場(chǎng)的�,是很有幫助的���!
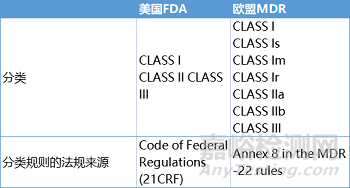
1���、分類
根據(jù)對(duì)病人和/或最終用戶影響的嚴(yán)重程度���,每種醫(yī)療器械都被劃分為一定的風(fēng)險(xiǎn)等級(jí)。每個(gè)等級(jí)都有不同的風(fēng)險(xiǎn)和相應(yīng)的損害效應(yīng)���。
美國(guó)FDA和歐盟的《醫(yī)療器械注冊(cè)和登記條例》都規(guī)定了醫(yī)療器械的三個(gè)基本等級(jí)�����。醫(yī)療器械分為低風(fēng)險(xiǎn)(I 級(jí))�����、中等風(fēng)險(xiǎn)(II 級(jí))和高風(fēng)險(xiǎn)(III 級(jí))�。在歐盟 MDR 中�����,還有一些更詳細(xì)的子分類���。
歐盟MDR的醫(yī)療器械根據(jù)MDR Annex 8中規(guī)定的規(guī)則進(jìn)行分類�����,其中有22條規(guī)則���,所有醫(yī)療器械都能在這些描述中找到�,針對(duì)醫(yī)療器械分類���,MDCG小組還發(fā)布了MDCG 2021-24“關(guān)于醫(yī)療器械分類的指導(dǎo)意見”�����。例外情況是�,隱形眼鏡等產(chǎn)品以及美容和紋身沙龍的某些器械也受到MDR法規(guī)的監(jiān)管���,盡管它們沒有嚴(yán)格定義的醫(yī)療目的。Annex 16提供了關(guān)于非醫(yī)療用途器械的更多信息�����。
在美國(guó)市場(chǎng)�,大多數(shù)醫(yī)療器械都可以通過21CFR Parts 862-892 中找到與器械相符的描述,對(duì)器械進(jìn)行分類�����。
如果一款新產(chǎn)品可以在以上法規(guī)部分中找到產(chǎn)品分類,那么它可以通過上市前通知(510(k))���,也可以通過上市前批準(zhǔn)(PMA)���。如果一款新產(chǎn)品不能根據(jù)21CFR 862-892進(jìn)行分類,則自動(dòng)被分為III類�,并必須通過上市前批準(zhǔn)。
510(k) 簡(jiǎn)單來說就是依據(jù)已有同類產(chǎn)品獲得批準(zhǔn)�����。這意味著制造商的醫(yī)療器械與市場(chǎng)上已有的另一種醫(yī)療器械相似�。其基本理念是,如果可以證明等同性���,那么產(chǎn)品就與同類產(chǎn)品一樣安全有效�����。FDA 必須審查 510(k) 并 "批準(zhǔn) "�,然后制造商才能在美國(guó)合法銷售或分銷該產(chǎn)品���,或在美國(guó)銷售���。
如果產(chǎn)品屬于 I 類或 II 類�����,則適用 510(k)�。根據(jù) 21CFR Part 814的規(guī)定�,III 類的醫(yī)療器械必須通過 PMA 。PMA 是一種監(jiān)管和科學(xué)程序�,用于驗(yàn)證醫(yī)療器械的安全性和有效性。上市前批準(zhǔn)的基礎(chǔ)是���,產(chǎn)品的安全性和有效性在任何時(shí)候都有足夠的臨床研究和科學(xué)證據(jù)作保證���。如果醫(yī)療器械因尚未上市而無法與任何上游器械進(jìn)行比較,則該器械將自動(dòng)被歸入III類�。
2、審批流程
審批流程是指醫(yī)療器械獲準(zhǔn)投放市場(chǎng)的流程�,本文為廣大制造商制作了審批流程差異表�����,詳細(xì)分解歐盟MDR和美國(guó)FDA的審評(píng)差異。
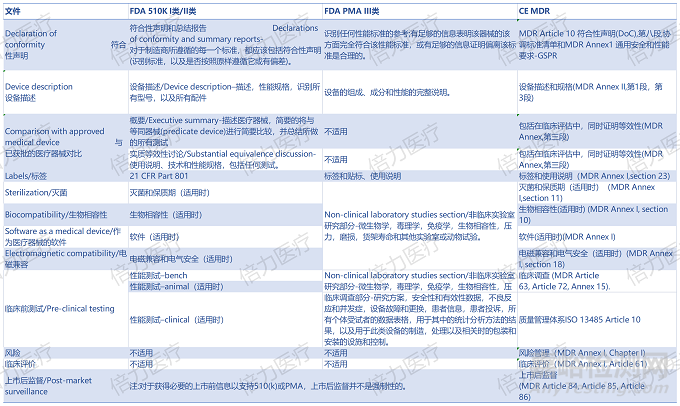
3�、技術(shù)文檔
在沒有系統(tǒng)性學(xué)習(xí)美國(guó)和歐盟的醫(yī)療器械法規(guī)時(shí),大部分人只是對(duì)FDA和CE的大致關(guān)鍵要素有些了解�����,比如:510K的本質(zhì)是實(shí)質(zhì)等同�����、CE-MDR的法規(guī)本質(zhì)是滿足法規(guī)的要求�����,臨床數(shù)據(jù)可以選擇等同器械的臨床文獻(xiàn)�����。那如何參照法規(guī)�����?如何搞懂全套技術(shù)文檔到底要什么�����?
注:本表只考慮與醫(yī)療器械直接相關(guān)的文件,不包括申請(qǐng) FDA 和公告機(jī)構(gòu)所需的任何行政文件�。
FDA和CE的異同總結(jié)
大多數(shù)經(jīng)FDA批準(zhǔn)的醫(yī)療器械都通過510K路徑獲得上市資格,除了與等同器械對(duì)比外�,大部分情況下是不用提供臨床信息的。因此這是和CE-MDR的最大區(qū)別�,在MDR法規(guī)下制造商將在根據(jù)MDR準(zhǔn)備臨床評(píng)估方面付出很大努力。
第二大區(qū)別是�,上市后監(jiān)督不需要獲得FDA的批準(zhǔn)。因此�,如果這是第一次在FDA注冊(cè)醫(yī)療器械,那么制造商沒有建立上市后監(jiān)督系統(tǒng)�����,而這對(duì)歐盟市場(chǎng)是強(qiáng)制性的���。對(duì)于在美國(guó)的公司�,他們的產(chǎn)品已經(jīng)在市場(chǎng)上上市一段時(shí)間(II類和II類)�,根據(jù)FDA的section 522 ,建立了a postmarket system���。