根據(jù)CE-MDR法規(guī)的要求��,所有醫(yī)療器械都必須進(jìn)行最新技術(shù)水平(SOTA)文獻(xiàn)審查�,從低風(fēng)險的I類到高風(fēng)險的III類有源植入器械�����。SOTA文獻(xiàn)綜述是醫(yī)療器械臨床評價過程的一部分���,它被定義為與器械相關(guān)的臨床數(shù)據(jù)的持續(xù)收集��、生成�、評估和分析。這一系統(tǒng)性文獻(xiàn)綜述的輸出為臨床評估�����、風(fēng)險管理和使用說明提供了信息����。它還為臨床調(diào)查的設(shè)計和設(shè)置提供了相關(guān)的輸入,以確保收集足夠的臨床證據(jù)來證明符合通用安全和性能要求(GSPR)和利益-風(fēng)險可接受標(biāo)準(zhǔn)����。
全面���、客觀和徹底的系統(tǒng)文獻(xiàn)綜述�����,以描述一般SOTA并確定評估器械(和/或等同器械)的所有相關(guān)臨床安全性����、性能和可用性數(shù)據(jù),應(yīng)符合歐盟MDR法規(guī)(EU) 2017/745和MEDDEV 2.7/1 Rev. 4的嚴(yán)格監(jiān)管指南����。本文帶您了解SOTA文獻(xiàn)搜索的不同步驟���。
1.SOTA文獻(xiàn)綜述-是什么��?為什么?要在什么情況下進(jìn)行�����?
系統(tǒng)性文獻(xiàn)綜述是一個結(jié)構(gòu)化和客觀的過程�����,旨在識別��、批判性評價���、分析和總結(jié)臨床證據(jù)����,以回答預(yù)先設(shè)定的研究問題。盡管在EU MDR 2017/745和MEDDEV 2.7/1 Rev. 4中多次提到SOTA����,但該術(shù)語本身沒有定義�����。SOTA的定義可在“MDCG 2020-6遺留器械臨床證據(jù)”中找到:“當(dāng)前技術(shù)能力和/或在產(chǎn)品、流程和患者管理方面接受的臨床實踐的發(fā)展階段�,基于相關(guān)的科學(xué)���、技術(shù)和經(jīng)驗的綜合發(fā)現(xiàn)����。注:最新技術(shù)水平體現(xiàn)了目前和普遍接受的技術(shù)和醫(yī)學(xué)的良好實踐���。最新技術(shù)水平并不一定意味著技術(shù)最先進(jìn)的解決方案��。”
根據(jù)MEDDEV 2.7/1 Rev. 4, SOTA文獻(xiàn)檢索和綜述應(yīng)建立:
1.General SOTA:概述醫(yī)療領(lǐng)域�����,技術(shù)�����,替代治療,和類似的基準(zhǔn)器械
2.被評估的器械和等同器械的臨床數(shù)據(jù)��,如果聲稱等同
由于SOTA文獻(xiàn)綜述是臨床評估過程中至關(guān)重要的第一步�����,理想情況下�����,在醫(yī)療器械的開發(fā)和上市前階段就已經(jīng)開始了。在此階段�����,SOTA文獻(xiàn)綜述為器械的醫(yī)療狀況和使用設(shè)定場景���,并檢索有關(guān)安全性����、性能和可用性參數(shù)的相關(guān)信息����,作為器械應(yīng)遵守的基準(zhǔn)��。系統(tǒng)的文獻(xiàn)回顧還確定了臨床獲益�����,并定義了器械的風(fēng)險-收益比概況的可接受標(biāo)準(zhǔn)。最初的SOTA文獻(xiàn)綜述的輸出是確定要生成哪些臨床數(shù)據(jù)以證明器械低估符合公認(rèn)的最新技術(shù)水平的關(guān)鍵��,從而確定臨床研究中應(yīng)該評估哪些臨床益處���、安全性、性能和可用性終點�,特別是在確定臨床證據(jù)差距的情況下。
最后�,SOTA文獻(xiàn)綜述為風(fēng)險管理提供了相關(guān)的輸入,因為它可以識別與器械相關(guān)的潛在臨床危害�,并通過提供有關(guān)危害頻率和嚴(yán)重程度的信息,幫助對危險情況進(jìn)行風(fēng)險評估�����。
為了獲得CE認(rèn)證�,與正在評估的器械有關(guān)的臨床證據(jù)應(yīng)具有足夠的數(shù)量和質(zhì)量,以確認(rèn)符合相關(guān)的GSPR和風(fēng)險-收益比概況的可接受標(biāo)準(zhǔn)����。對于某些醫(yī)療器械,特別是I類和IIa類器械�����,通過文獻(xiàn)綜述產(chǎn)生的臨床數(shù)據(jù)可能代表了大部分(如果不是全部)臨床證據(jù)(由于缺乏臨床調(diào)查)�,強(qiáng)調(diào)了進(jìn)行良好和可靠的系統(tǒng)文獻(xiàn)綜述的相關(guān)性�����。
2.SOTA文獻(xiàn)綜述-更新
在器械的整個生命周期中���,臨床評估及其文件應(yīng)更新從上市后市場監(jiān)督(PMS)和上市后臨床跟蹤(PMCF)獲得的臨床數(shù)據(jù)�,以便在投放市場后主動收集和評估器械的安全性和性能數(shù)據(jù)���。
因此,在獲得CE認(rèn)證后�����,作為PMS的一部分����,SOTA文獻(xiàn)綜述將繼續(xù)進(jìn)行�,為了:
i) 跟上醫(yī)療領(lǐng)域的當(dāng)前知識���;
ii) 持續(xù)監(jiān)測器械的安全性����、性能��、可用性和風(fēng)險-收益比概況���;
iii) 跟蹤任何不可預(yù)見的風(fēng)險或器械誤用�����。
SOTA和臨床評估更新的頻率取決于制造商的器械的風(fēng)險分類和PMS的輸出:
·每當(dāng)PMS的新信息影響臨床評估或其結(jié)論時(考慮所有風(fēng)險類別),特別是器械的風(fēng)險-收益比概況
·如果沒有收到新的信息�����,那么:
a.對于高風(fēng)險(III類)或尚未完善的器械��,至少每年檢查一次
b.對于低風(fēng)險和建立良好的器械,每2-5年一次����。
定期對評估中的器械進(jìn)行文獻(xiàn)回顧更新,確保領(lǐng)先于新出現(xiàn)的風(fēng)險���,并允許其在風(fēng)險出現(xiàn)時升級和減輕風(fēng)險。主動識別風(fēng)險允許采取保護(hù)措施并將風(fēng)險降低到可接受的水平���。
3.SOTA文獻(xiàn)綜述-過程和文件
為了計劃和記錄文獻(xiàn)綜述的搜索和輸出�,設(shè)計良好且書面清晰的literature review plan or protocol (LRP) and report (LRR) /文獻(xiàn)綜述計劃或方案(LRP)和報告(LRR)是所有器械分類(包括所有新器械和遺留器械)的關(guān)鍵和必需的��。
LRP和LRR作為醫(yī)療器械臨床證據(jù)和技術(shù)文件的一部分����,是NB審查的兩個關(guān)鍵文件�,兩份文件都應(yīng)注明日期,版本控制�����,并由法規(guī)制定者��、評估者和制造商簽署�����。
3.1 文獻(xiàn)綜述方案
文獻(xiàn)綜述始于制定文獻(xiàn)綜述計劃或方案,該計劃或方案應(yīng)描述文獻(xiàn)綜述的背景和范圍����。文獻(xiàn)綜述的基本原理和器械的描述,其預(yù)期用途,適應(yīng)癥�����,目標(biāo)人群和用戶包括在本文檔中�����。該方案還詳細(xì)介紹了相關(guān)出版物的識別�、選擇和評價方法��,以解決研究問題��。
文獻(xiàn)檢索方法學(xué)應(yīng)在LRP中透明����、正確地記錄���,以保證檢索可以重復(fù),方法可以得到嚴(yán)格評價�����,結(jié)果可以得到驗證。
3.1.1 文獻(xiàn)檢索策略
搜索策略應(yīng)該是徹底和客觀的����,并確定所有相關(guān)的有利和不利的數(shù)據(jù)�����。SOTA文獻(xiàn)綜述與評估器械的預(yù)期用途����、適應(yīng)癥����、目標(biāo)人群和性能有關(guān),并應(yīng)包括:
·General SOTA���,包括醫(yī)療領(lǐng)域的臨床實踐指南
·類似基準(zhǔn)器械/替代治療的安全性、性能和可用性數(shù)據(jù)
·被評估的器械和/或等同器械的安全性�、性能和可用性數(shù)據(jù)(如果聲稱等同)
我們優(yōu)先應(yīng)用不同來源的臨床文獻(xiàn)。為了獲得必要的信息和數(shù)據(jù)��,通常需要使用不同焦點�����、搜索條件和篩選器進(jìn)行多次搜索�。MEDLINE (Pubmed)是最常用的搜索引擎���。
為了確保歐洲器械和治療的充分覆蓋����,并識別所有使用評估器械和類似基準(zhǔn)器械進(jìn)行的研究����,還指定了替代數(shù)據(jù)庫�����,如但不限于EMBASE/COCHRANE/Google Scholar�����。如需搜索各自醫(yī)療領(lǐng)域的現(xiàn)行實踐指南,可以使用TRIP和UpToDate等更具體的數(shù)據(jù)庫���。在適當(dāng)?shù)那闆r下��,可以另外應(yīng)用手工網(wǎng)絡(luò)搜索����。此外�����,考慮咨詢類似競爭對手器械的使用說明���,并篩選通過不同搜索檢索到的相關(guān)出版物的參考書目。應(yīng)提供選擇相應(yīng)數(shù)據(jù)庫的理由
通過文獻(xiàn)回顧����,采用無偏倚��、系統(tǒng)��、檢索的方法來制定待回答的研究問題���?��;赑ICO的搜索策略是一種被普遍接受的���、基于證據(jù)的方法����,用于設(shè)置覆蓋評估器械的SOTA文獻(xiàn)所需的不同搜索��。PICO代表人群����、干預(yù)��、比較和結(jié)局����。通過制定與每個PICO類別相關(guān)的研究問題�����,生成相關(guān)關(guān)鍵詞����,用于搜索的設(shè)置��。
在生成相關(guān)的關(guān)鍵字之后����,還需要為不同的搜索建立搜索字符串����。正確使用布爾邏輯/boolean logic(AND、OR���、NOT)是關(guān)鍵,應(yīng)用特定的過濾器(如出版物類型和日期范圍)有助于集中搜索��。應(yīng)在計劃中說明應(yīng)用任何納入和排除標(biāo)準(zhǔn)���、限制和篩選器的基本原理。
3.1.2 篩選和選擇過程
在不同的數(shù)據(jù)庫中進(jìn)行系統(tǒng)搜索后��,應(yīng)記錄和跟蹤搜索輸出����,并由參考管理器軟件提供便利�����。在刪除重復(fù)記錄后�����,通常應(yīng)用一個逐步選擇過程。首先根據(jù)標(biāo)題和摘要對引文進(jìn)行篩選�,如果評估為可能相關(guān),則對全文出版物進(jìn)行篩選��,以獲得相關(guān)SOTA��、安全性和性能數(shù)據(jù)。相關(guān)文獻(xiàn)的選擇應(yīng)客觀���,排除記錄的理由應(yīng)適當(dāng)記錄。在文獻(xiàn)綜述報告中����,采用流程圖將篩選和選擇過程可視化。最終被選擇納入文獻(xiàn)綜述的出版物����,也被篩選在參考書目中引用的有趣的引文�����。
3.1.3 數(shù)據(jù)分析與評價
為了描述general SOTA�����,許多數(shù)據(jù)和信息來自綜述文章、meta分析和指南����,并輔以原始研究論文的數(shù)據(jù)。
為了評估被評估器械����、類似競爭對手器械和其他替代治療的安全性�����、性能和可用性,與這些結(jié)果參數(shù)相關(guān)的數(shù)據(jù)應(yīng)從原始研究出版物中檢索����。對于每個數(shù)據(jù)集�����,一般研究特征��,如研究設(shè)計�����、研究人群和研究器械���,以及定量和/或定性的安全性、性能和可用性數(shù)據(jù)被提取并呈現(xiàn)在數(shù)據(jù)提取表中���。
從出版物中提取的臨床安全性、性能和可用性數(shù)據(jù)需要進(jìn)行分析和評估。根據(jù)MDR法規(guī)(EU) 2017/745和MEDDEV 2.7/1 Rev. 4的Annex XIV�,所有與評估器械相關(guān)的臨床數(shù)據(jù)都應(yīng)通過評估其建立器械安全性和性能的適用性來進(jìn)行評估。每個單獨的數(shù)據(jù)集應(yīng)該在科學(xué)有效性���、相關(guān)性、質(zhì)量和對臨床安全性��、性能和/或可用性的貢獻(xiàn)方面進(jìn)行評估���。為了確保系統(tǒng)�����、透明和客觀的評估����,數(shù)據(jù)評估的方法和標(biāo)準(zhǔn)應(yīng)該在文獻(xiàn)綜述計劃中預(yù)先設(shè)定和證明��。
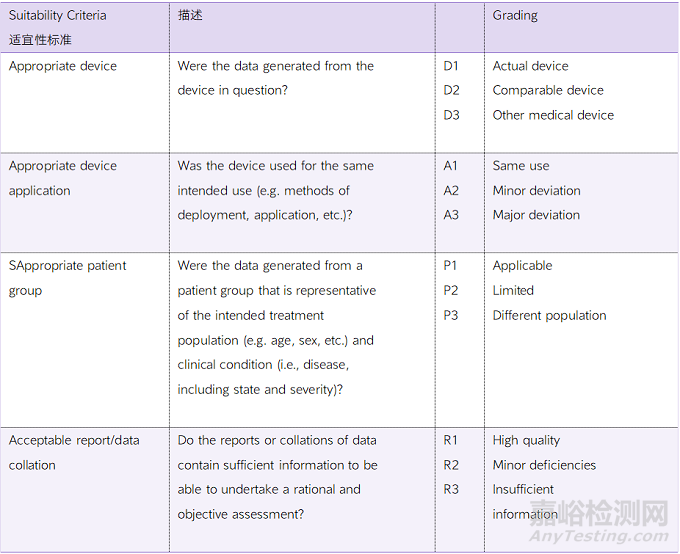
適當(dāng)和一致地權(quán)衡有利和不利的數(shù)據(jù)是重要的���。歐盟MDR法規(guī)(EU) 2017/745和MEDDEV 2.7/1 Rev. 4規(guī)定了一個明確的削減,完善的評估方法。在IMDRF MDCE WG/N56FINAL:2019中����,通過一個基于數(shù)據(jù)適宜性和數(shù)據(jù)貢獻(xiàn)標(biāo)準(zhǔn)的示例提供了指導(dǎo)(見下圖)。這些評估標(biāo)準(zhǔn)的應(yīng)用決定了每個單獨數(shù)據(jù)集的證據(jù)權(quán)重���,這對于總體數(shù)據(jù)分析和制定與器械安全性和性能相關(guān)的總體結(jié)論至關(guān)重要。
表1-適宜性評估標(biāo)準(zhǔn)樣本
(來源:IMDRF MDCE WG/N56FINAL:2019)
表2-數(shù)據(jù)貢獻(xiàn)樣本評估標(biāo)準(zhǔn)
(來源:IMDRF MDCE WG/N56FINAL:2019)
3.2 文獻(xiàn)綜述報告
文獻(xiàn)檢索和綜述的輸出在文獻(xiàn)綜述報告中進(jìn)行描述和總結(jié)�,該報告包含對正在評估的器械和/或等同器械(如果申請)和類似基準(zhǔn)器械的當(dāng)前知識/最新技術(shù)水平和檢索到的臨床數(shù)據(jù)的綜述��。
LRR的正文一方面包括對最新技術(shù)水平的描述���,另一方面包括對與器械和類似器械有關(guān)的已發(fā)表的臨床安全性、性能和可用性數(shù)據(jù)的系統(tǒng)分析�。所有數(shù)據(jù)集應(yīng)被記錄����、充分分析、評估����、總結(jié),并在LRR中引用����。
3.2.1 General SOTA
MEDDEV 2.7/1 Rev. 4 appendix A9對SOTA部分的內(nèi)容提供了指導(dǎo)。SOTA定義了被評估器械的臨床背景�、使用器械的醫(yī)療領(lǐng)域和目標(biāo)人群��。應(yīng)確定疾病的自然病程�、不同階段、發(fā)病率�����、嚴(yán)重程度和后果。此外���,應(yīng)該討論不同的替代治療方案和具有每種替代方案(dis)優(yōu)勢的類似競爭器械����。根據(jù)MDR法規(guī)(EU) 2017/745�,對相同適應(yīng)癥的替代治療方法的審查越來越嚴(yán)格。應(yīng)詳細(xì)說明所評估的器械和文獻(xiàn)中描述的類似基準(zhǔn)器械和/或替代治療的益處和風(fēng)險�����。
本節(jié)還介紹了用于預(yù)期醫(yī)療目的的器械的臨床實踐指南和建議����。最后�����,General SOTA概述了未滿足的醫(yī)療需求和器械的用戶類型�����。
為了檢索General SOTA內(nèi)容��,應(yīng)用一個更高級的搜索字符串�,該字符串使用涵蓋醫(yī)療狀況和適應(yīng)癥(可能包括通用器械組和替代療法)的搜索詞�。本檢索集中于系統(tǒng)綜述、薈萃分析和臨床實踐指南�。通過這些出版物類型檢索到的信息豐富了來自原始研究出版物或所選綜述文章中引用的出版物的數(shù)據(jù)�����。所涵蓋的時間段最好至少從搜索日期算起10年。
3.2.2 同類器械/替代治療的臨床資料
鑒于臨床評估�,重要的是要記住�����,應(yīng)將評估中的器械與基準(zhǔn)器械進(jìn)行比較����,并根據(jù)現(xiàn)有的治療選擇和/或針對預(yù)期醫(yī)學(xué)適應(yīng)癥的可用器械討論其風(fēng)險、性能和臨床益處��。
系統(tǒng)文獻(xiàn)綜述的目標(biāo)之一是獲得所研究器械的安全性和性能可接受性標(biāo)準(zhǔn)�。為此,最好在文獻(xiàn)綜述計劃中識別并預(yù)先列出類似的器械;
并在文獻(xiàn)綜述報告中對市場上同類競爭對手器械的數(shù)據(jù)進(jìn)行提取���、分析和總結(jié)。相似器械是指屬于同一通用器械組的設(shè)備�����。
MDR Article 2(7)將其定義為具有相同或類似預(yù)期目的或技術(shù)共性的一組器械��,允許它們以不反映特定特征的通用方式進(jìn)行分類����。術(shù)語“同類器械”應(yīng)與“等同器械”明確區(qū)分��,后者是具有相同技術(shù)���、生物學(xué)和臨床特征的器械,其安全性和臨床性能在臨床上不會有顯著差異(MDR Annex XIV Part a)�。如果市場上沒有類似的競爭對手器械,或者競爭對手器械的安全性和性能數(shù)據(jù)不足�,在臨床評價過程中,可提取備選SOTA治療方案的數(shù)據(jù)��,優(yōu)選器械與待評價器械進(jìn)行比較�。例如����,備選器械可以涉及具有相同預(yù)期用途但不同適應(yīng)癥、患者群體或作用方式的器械��。
同類基準(zhǔn)器械或替代品的安全性����、性能和可用性數(shù)據(jù)是從已發(fā)表的原始臨床研究中提取的。為了獲得這些數(shù)據(jù)��,更具體的搜索字符串使用涵蓋適應(yīng)癥�、器械組和已識別的類似器械或替代方案的搜索術(shù)語,可能包括已確定的性能和安全結(jié)果�。搜索周期取決于正在評估的器械的監(jiān)管狀態(tài)���。對于遺留器械�����,CE認(rèn)證的日期通常被選擇為開始日期,除非之前進(jìn)行了綜合文獻(xiàn)審查或基準(zhǔn)器械已在市場上上市多年�。對于新器械的初始CE認(rèn)證標(biāo)志,所涵蓋的期間從同類器械(或如果沒有類似器械銷售的替代品)在市場上可用時開始�����。
通過對已發(fā)表文獻(xiàn)的全文綜述提取定量和定性的安全性和性能數(shù)據(jù)���。在專用的數(shù)據(jù)提取表中輸入的數(shù)據(jù)應(yīng)進(jìn)行分析和匯總��,最好在文本匯總表中使用,在報告中��。適用時����,安全性和性能總結(jié)應(yīng)按適應(yīng)癥�����、器械類型和隨訪(至少對于植入器械)提供����。在不同的摘要中應(yīng)說明研究的數(shù)量(包括數(shù)據(jù)集的評估和權(quán)重)����,以及對安全性和性能數(shù)據(jù)做出貢獻(xiàn)的患者數(shù)量,以便對文獻(xiàn)數(shù)據(jù)進(jìn)行適當(dāng)?shù)目傮w解讀�。
3.2.3 待評器械/等同器械的臨床資料
最后但并非最不重要的是�����,應(yīng)該建立檢索非制造商生成的正在評估的器械(以及等同器械�����,如果適用)的安全性,性能和可用性數(shù)據(jù)的搜索�����。作為CER中PMCF數(shù)據(jù)收集的一部分��,通常在CE后進(jìn)行與評估器械相關(guān)的臨床文獻(xiàn)搜索��。顯然���,要獲得這些數(shù)據(jù),需要編譯包含器械商品名稱和制造商的單獨搜索字符串�����。數(shù)據(jù)提取��、評估和分析的執(zhí)行方式與類似競爭對手的器械/替代療法相同����。
3.2.4 文獻(xiàn)綜述的結(jié)論
文獻(xiàn)綜述應(yīng)得出關(guān)于評估器械的安全性和性能的明確結(jié)論����,并討論與類似基準(zhǔn)器械或替代治療方案相關(guān)的結(jié)果�。確定證據(jù)中的任何空白,以確認(rèn)在器械預(yù)期使用的正常條件下符合GSPR���。評估臨床文獻(xiàn)是否支持說明書中所述的預(yù)期目的�,以及所評估器械的任何益處和臨床性能聲明��。
SOTA文獻(xiàn)綜述的輸出是醫(yī)療器械的風(fēng)險管理和臨床評價����。它提供了被評估器械的醫(yī)療背景��,確定了類似的基準(zhǔn)器械和/或替代治療方案��,并規(guī)定了器械安全性��、性能和可用性以及收益-風(fēng)險規(guī)格的可接受標(biāo)準(zhǔn)。
4. 文獻(xiàn)綜述-誰來寫����?
文獻(xiàn)綜述優(yōu)先由具有科學(xué)或醫(yī)學(xué)背景���、了解研究方法學(xué)、信息管理和法規(guī)要求的合格醫(yī)學(xué)作者進(jìn)行�。強(qiáng)烈建議臨床���、質(zhì)量和監(jiān)管專家作為LRP和LRR的評估者提供意見����。根據(jù)MEDDEV 2.7/1 rev 4對LRP和LRR作者和評估者的專業(yè)知識和經(jīng)驗的具體要求��,包括相關(guān)的高等教育學(xué)位和5年相關(guān)的專業(yè)經(jīng)驗�����,或10年的專業(yè)經(jīng)驗(如果學(xué)位不被認(rèn)為是任務(wù)的先決條件)���。報告中應(yīng)提供所有作者和評估者的最新(截至)簡歷。與上述要求的偏差應(yīng)被記錄并適當(dāng)?shù)刈C明����。所有評估者必須提供利益聲明。
5. 文獻(xiàn)綜述-常見的差距和障礙
5.1 文獻(xiàn)綜述不佳
糟糕的文獻(xiàn)檢索策略和方法是臨床評估中的一個常見“差距”�。不恰當(dāng)?shù)臄?shù)據(jù)庫選擇�����,不充分的檢索周期�����,不充分的檢索術(shù)語���,缺乏在和排除標(biāo)準(zhǔn)的理由,SOTA評估和臨床數(shù)據(jù)的權(quán)衡導(dǎo)致不合規(guī)并危及您的醫(yī)療器械的CE標(biāo)志�����。請記住����,一個完善的文獻(xiàn)綜述計劃可以很容易地重復(fù),并將在更新CER時為您節(jié)省大量時間����。
5.2 關(guān)鍵文件不一致
一個常見的缺陷是信息材料(IFU��、用戶手冊、風(fēng)險管理文件)���、文獻(xiàn)綜述和臨床評估文件之間的不一致�,特別是關(guān)于器械聲明�。確保CE提交技術(shù)文件中的所有文件與預(yù)期用途、適應(yīng)證����、患者人群以及安全性��、性能和臨床效益聲明相一致���。
5.3 來自臨床專家的意見
SOTA文獻(xiàn)綜述的執(zhí)行以及LRP和LRR的實際撰寫通常是醫(yī)學(xué)法規(guī)作者的一項專門任務(wù)。根據(jù)現(xiàn)行法規(guī)和要求的高標(biāo)準(zhǔn)建立扎實的文獻(xiàn)檢索�����,強(qiáng)烈建議由相關(guān)醫(yī)學(xué)領(lǐng)域的臨床專家參與����。專家的輸入、反饋和建議是相關(guān)關(guān)鍵詞選擇和納入��、排除標(biāo)準(zhǔn)建立的關(guān)鍵��。在建立搜索之前�����,對SOTA范圍的公開討論和明確溝通是快速準(zhǔn)備好LRP和LRR文檔的核心���。
5.4 低估時間和預(yù)算
文獻(xiàn)綜述的準(zhǔn)備和執(zhí)行需要時間���。然而�,精心策劃的文獻(xiàn)檢索將允許您從一開始就正確地報告SOTA�,并在醫(yī)療器械的整個生命周期中輕松復(fù)制文獻(xiàn)綜述更新���。定性的SOTA文獻(xiàn)回顧是必不可少的��,將為有效的臨床評估器械鋪平道路�����。在計劃階段����,與器械專家密切合作���,仔細(xì)定義搜索并編寫LRP至少需要25至30小時����。在搜索的計劃和設(shè)置上投入時間可以防止你不得不一遍又一遍地做這些工作��,最終肯定會有回報的�����。從不同檢索中篩選和選擇產(chǎn)出��,以及對相關(guān)出版物進(jìn)行數(shù)據(jù)提取和評價所需的時間��,高度依賴于產(chǎn)出的點擊量和特定醫(yī)學(xué)領(lǐng)域發(fā)表的相關(guān)研究的數(shù)量����。
對文獻(xiàn)數(shù)據(jù)的分析和綜合以及文獻(xiàn)綜述報告的實際撰寫預(yù)計還需要30-40個小時����。從開始到結(jié)束的整個過程,包括醫(yī)學(xué)作者�、臨床專家和制造商之間的交互,可能需要長達(dá)2-3個月����。
5.5 模板符合MDR 和meddev 2.7/1 rev 4
對于旨在獲得醫(yī)療器械認(rèn)證的初創(chuàng)公司來說����,LRP和LRR及其相關(guān)臨床文件的準(zhǔn)備可能是一項艱巨的任務(wù)。為了獲得CE批準(zhǔn)����,基本文件應(yīng)符合適用法規(guī)����,然而�����,符合MDR 和MEDDEV 2.7/1 rev 4的系統(tǒng)文獻(xiàn)和臨床評價文件的模板不容易從歐盟官方來源獲得��。