質(zhì)量研究的目的是通過對影響藥品質(zhì)量的各方面因素進(jìn)行研究�����,確定控制藥品質(zhì)量的項目、分析方法�,并綜合藥學(xué)研究、藥理毒理和臨床研究的結(jié)果制定終產(chǎn)品的質(zhì)量標(biāo)準(zhǔn)�,主要關(guān)注以下幾方面:
1.手性雜質(zhì)的確定
在手性藥物質(zhì)量研究中的一個關(guān)鍵問題就是對藥物光學(xué)純度的控制。而影響藥物光學(xué)純度的手性雜質(zhì)主要來源于兩個方面:原料藥的制備工藝中引入的手性原料���、手性試劑���、反應(yīng)副產(chǎn)物及副反應(yīng)產(chǎn)物等工藝雜質(zhì);原料藥本身不穩(wěn)定,構(gòu)型發(fā)生翻轉(zhuǎn)而形成的立體異構(gòu)體���。因此在研究之初, 就需根據(jù)原料藥的制備工藝與各手性中心的穩(wěn)定性確定需要控制的手性雜質(zhì)���。在這方面存在的一個主要誤區(qū)即是:當(dāng)分子中存在多個手性中心時, 不考慮制備工藝與各手性中心的穩(wěn)定性情況,不加區(qū)分地只對并不可能存在的對映異構(gòu)體雜質(zhì)進(jìn)行研究控制, 而對實際可能會存在的非對映異構(gòu)體雜質(zhì)卻毫無分析與研究。
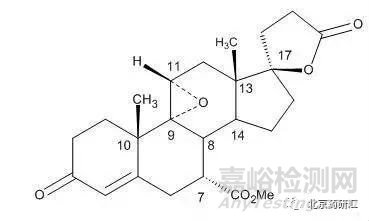
實例1:依普利酮分子中存在8個手性中心�,結(jié)構(gòu)如下:
由結(jié)構(gòu)式可知:其中有些手性中心,如8、10�、13位是處于甾體母核上,一般是天然形成的剛性結(jié)構(gòu),不太容易發(fā)生構(gòu)型的翻轉(zhuǎn);而有些手性中心�,如7、9、11���、17則大都是后續(xù)反應(yīng)中引入的,可能會產(chǎn)生相應(yīng)的立體異構(gòu)體雜質(zhì)�。因此,該藥物中可能存在的立體異構(gòu)體雜質(zhì)只可能是非對映異構(gòu)體,而不可能存在對映異構(gòu)體雜質(zhì)�����。
2.方法的選擇及驗證
目前常用于控制手性藥物光學(xué)純度的方法有兩種:比旋度法與手性色譜法���。影響比旋度數(shù)值的因素較多, 該數(shù)值的變化并不一定能靈敏、準(zhǔn)確地反映出立體異構(gòu)體含量的變化, 當(dāng)比旋度數(shù)值較小時更是如此�����。例如,當(dāng)采用1 dm的旋光管, 濃度為1g·100mL-1的溶液進(jìn)行測定時,假設(shè)某手性藥物僅含有一個手性中心, 該藥物的比旋度為+100o�。則根據(jù)計算公式可知:當(dāng)該藥物中混有1.0%的左旋異構(gòu)體雜質(zhì)(假設(shè)其他雜質(zhì)忽略不計)時,此時藥物的旋光度將由+1.00o變?yōu)?0.98o而如果該手性藥物的比旋度僅為+10o, 則當(dāng)該藥物中同樣混有1.0%的左旋異構(gòu)體雜質(zhì)時,此時藥物的旋光度將由+0.10o變?yōu)?0.098o, 旋光度的變化值僅為0.002o。此時如仍采用藥典附錄中提到的讀數(shù)至0.01o的旋光計, 則兩者旋光度的差值超出了儀器的測量精度, 在該旋光計上將顯示相同的讀數(shù), 根本反映不出產(chǎn)品中混有1.0%的左旋異構(gòu)體雜質(zhì)�。這也是為什么在制定質(zhì)量標(biāo)準(zhǔn)時,當(dāng)手性藥物在不同溶劑中的比旋度相差較大時, 一般選擇具有較大比旋度數(shù)值的溶劑作為標(biāo)準(zhǔn)中測定用溶劑, 以盡可能靈敏地反映手性藥物光學(xué)純度的變化情況。
實例2:甲基多巴在0.1N HCl中的比旋度為[α]25D-5.2(C=2.0), 在水中的比旋度為[ α] 20 D-14(C=1.09), 而在4.4% AlCl3水溶液中的比旋度為-25~-28���。為保證該項目更能靈敏地反映甲基多巴光學(xué)純度的變化,在中國藥典收載的甲基多巴標(biāo)準(zhǔn)中,就采用了配置較復(fù)雜的4.4%AlCl3水溶液作為測定比旋度的溶劑, 而未采用常見的0.1N HCl或水作溶劑���。
另外,當(dāng)手性藥物含有多個手性中心時, 隨著立體異構(gòu)體數(shù)目的增多, 比旋度數(shù)值的變化將會受到更大的影響, 從而更加難以從比旋度的變化來獲知手性藥物光學(xué)純度的具體變化情況。
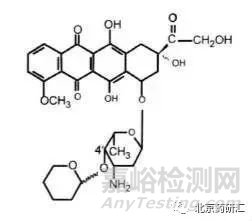
實例3:吡柔比星在4'位產(chǎn)生的2個差向異構(gòu)體在氯仿中的比旋度是相同的,均為+150(C=0.2)。此時如果僅用測定吡柔比星在氯仿中的比旋度來控制4'位差向異構(gòu)體的含量就變得毫無意義了�。因為不論該差向異構(gòu)體的含量如何變化,所測定的比旋度均不變。
基于以上原因,手性色譜法可以更為準(zhǔn)確直觀地反映各立體異構(gòu)體雜質(zhì)的變化情況���。但是,在質(zhì)量研究中同樣需要對所采用的手性色譜法進(jìn)行充分的方法學(xué)驗證, 以確保該方法確實能夠以足夠的靈敏度檢測到需控制的立體異構(gòu)體雜質(zhì)���。為此, 在驗證時需在以下兩方面加以注意:分析確定需控制的立體異構(gòu)體雜質(zhì);盡量采用雜質(zhì)對照品進(jìn)行專屬性和定量限的驗證�。在分析確定需控制的立體異構(gòu)體雜質(zhì)時, 首先應(yīng)根據(jù)原料藥的制備工藝, 分析工藝中可能產(chǎn)生哪些立體異構(gòu)體雜質(zhì);其次, 需要設(shè)計一系列的降解試驗來考察確定分子中各手性中心構(gòu)型的穩(wěn)定性, 以分析確定該手性藥物在哪些因素作用下可能降解產(chǎn)生哪些立體異構(gòu)體雜質(zhì)。這樣,在分析方法驗證時, 就能目的明確地考察該分析方法是否能分離并檢測出所有可能存在的立體異構(gòu)體雜質(zhì)���。
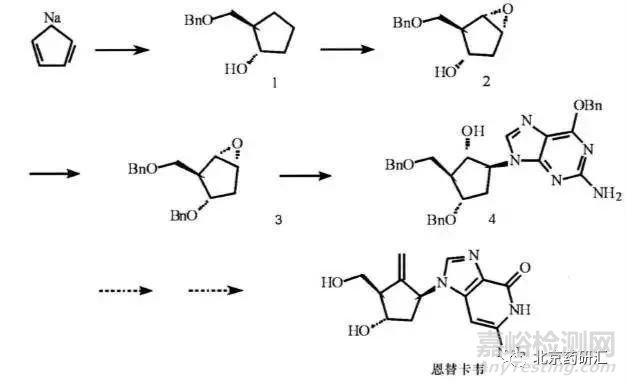
實例4:恩替卡韋
抗乙肝病毒藥物恩替卡韋的合成���,在引入手性中心的第1, 2, 4 步反應(yīng)中, 只要這些反應(yīng)不是采用立體專屬性的反應(yīng), 以百分之百地得到所需構(gòu)型的反應(yīng)產(chǎn)物, 且后續(xù)操作中不能有效地去除所產(chǎn)生的立體異構(gòu)體雜質(zhì), 則終產(chǎn)品中就可能混有恩替卡韋的所有其他7個立體異構(gòu)體雜質(zhì)。除非在反應(yīng)過程中已采用合適的純化手段去掉各步反應(yīng)中引入的立體異構(gòu)體雜質(zhì),并采用專屬有效的分析方法對各步反應(yīng)的中間體中的立體異構(gòu)體雜質(zhì)進(jìn)行監(jiān)控, 否則在終產(chǎn)品的質(zhì)量控制中,就應(yīng)采用合適的分析方法對這7個立體異構(gòu)體雜質(zhì)進(jìn)行控制�����。此時終產(chǎn)品的質(zhì)控難度是非常大的�。
由此例可見,了解手性藥物的制備工藝, 并通過對引入手性中心的化學(xué)反應(yīng)的立體選擇性進(jìn)行分析, 可以全面了解工藝中可能引入的立體異構(gòu)體雜質(zhì),從而采取合適的分析手段分別在工藝過程中或終產(chǎn)品中進(jìn)行控制質(zhì)量標(biāo)準(zhǔn)的制定,在制定手性藥物的質(zhì)量標(biāo)準(zhǔn)時一定要結(jié)合手性藥物的特點,重點關(guān)注能否切實控制手性藥物的光學(xué)純度���。對于合成制備的手性原料藥,由于合成過程中會引入各種手性雜質(zhì),并且在放置過程中也可能會因為構(gòu)型不穩(wěn)定而降解產(chǎn)生相應(yīng)的立體異構(gòu)體雜質(zhì),所以原料藥的質(zhì)量標(biāo)準(zhǔn)中一般均須設(shè)置專屬而靈敏的立體異構(gòu)體雜質(zhì)檢測項目���。
對于發(fā)酵或提取制備的手性原料藥, 由于各手性中心均是天然形成的,一般都是立體專屬性的,產(chǎn)生立體異構(gòu)體雜質(zhì)的可能性不大,如果有充足的文獻(xiàn)或?qū)嶒炓罁?jù)證明各手性中心在放置過程中構(gòu)型是穩(wěn)定的,則可以僅用比旋度項目來粗略控制該手性藥物的光學(xué)特性�。
對于制劑而言:如果手性原料藥在放置過程中構(gòu)型是穩(wěn)定的, 且原料藥中所含立體異構(gòu)體雜質(zhì)不具生理活性,則制劑的質(zhì)量標(biāo)準(zhǔn)中可不納入立體異構(gòu)體雜質(zhì)控制項目���。但是,隨著研究的深入,越來越多的已上市消旋體藥物被開發(fā)成單一的立體異構(gòu)體藥物�。
實例5:在苯磺酸氨氯地平片的基礎(chǔ)上進(jìn)一步開發(fā)了苯磺酸左旋氨氯地平片�。兩者的區(qū)別僅在于后者的規(guī)格較前者小一半。此時如果不根據(jù)苯磺酸左旋氨氯地平片的研究結(jié)果,在質(zhì)量標(biāo)準(zhǔn)中酌情制定立體異構(gòu)體雜質(zhì)檢查項或立體專屬性的鑒別項,則在質(zhì)量標(biāo)準(zhǔn)中很難區(qū)分消旋體與左旋體片劑�����。因此有必要在單一的立體異構(gòu)體制劑的質(zhì)量標(biāo)準(zhǔn)中至少訂入立體專屬性的鑒別項,以利于上市藥品的監(jiān)督管理�����。
3.穩(wěn)定性研究
手性藥物在一定條件下,手性中心可能會發(fā)生構(gòu)型反轉(zhuǎn),從而生產(chǎn)相應(yīng)的立體異構(gòu)體�。正是由于構(gòu)型有可能發(fā)生翻轉(zhuǎn),所以在手性藥物的穩(wěn)定性研究中就應(yīng)該采取有效的分析方法監(jiān)控各手性中心構(gòu)型的穩(wěn)定性�����。在此需注意以下幾方面的問題:
① 如前所述, 基于比旋度法的局限性, 最好采用更為靈敏的手性色譜法來監(jiān)測各個立體異構(gòu)體的變化情況�����。
② 要注意考察制劑中的手性藥物構(gòu)型是否仍然穩(wěn)定, 尤其是一些液體制劑。
③ 要根據(jù)手性中心的多寡與各自的穩(wěn)定性, 選擇合適的立體異構(gòu)體進(jìn)行檢測�。
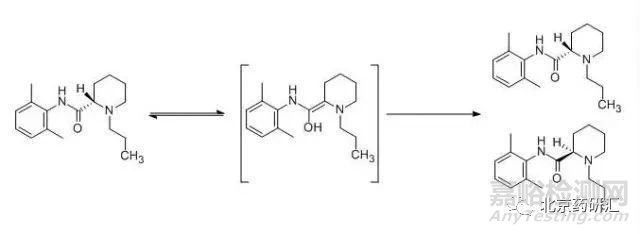
實例6:在長效酰胺類局麻藥--甲磺酸羅哌卡因的質(zhì)量標(biāo)準(zhǔn)中,為檢測手性HPLC法的系統(tǒng)適應(yīng)性,取甲磺酸羅哌卡因?qū)φ掌愤m量,加水制成每1mL含75μg的溶液,于90℃放置6h后,即可得到含有0.5%至1.0% R-(+)對映異構(gòu)體的對照溶液。由此可見,甲磺酸羅哌卡因分子中的手性中心在溶液中加熱即可發(fā)生構(gòu)型反轉(zhuǎn)而生成其對映異構(gòu)體�。
由羅哌卡因的結(jié)構(gòu)可知,手性中心的鄰位有吸電子的羰基,從而使手性中心上的氫成為活性氫,部分活性氫原子在溶液中獲取一定的能量即可以氫離子的形式脫去,使手性中心成為平面結(jié)構(gòu)的負(fù)碳離子,根據(jù)進(jìn)攻方向立體位阻的大小,溶液中的氫離子再以一定的比例從平面的上、下兩個方向進(jìn)攻該負(fù)碳離子,從而分別得到甲磺酸羅哌卡因及少量的右旋體雜質(zhì)�。
實例7:二氫膽固醇構(gòu)型的轉(zhuǎn)換
二氫膽固醇在堿的催化下, 其3位的α構(gòu)型的羥基會部分轉(zhuǎn)化為β 構(gòu)型,從而產(chǎn)生非對映異構(gòu)體�����。反應(yīng)示意圖見圖5�����。故在此情況下, 穩(wěn)定性考察時監(jiān)測的重點降解產(chǎn)物就應(yīng)該是非對映異構(gòu)體, 而不是對映異構(gòu)體�����。
此外,在考察手性藥物構(gòu)型的穩(wěn)定性時, 考察條件不能局限于國內(nèi)穩(wěn)定性研究指導(dǎo)原則通常所采用的影響因素試驗的條件�����。例如, 前面提到的甲磺酸羅哌卡因, 如采用一般的影響因素試驗條件, 就可能觀察不到構(gòu)型的翻轉(zhuǎn)現(xiàn)象�����。
綜上所述, 在手性藥物的藥學(xué)研究中, 一定要結(jié)合手性藥物的特點開展相應(yīng)的研究,否則所作的研究難以真正反映手性藥物的穩(wěn)定性,也難以準(zhǔn)確控制其光學(xué)純度。另外,在具體的研究過程中,也不能拘泥于指導(dǎo)原則中的方法機(jī)械地執(zhí)行�。而是要在遵循其基本原則的基礎(chǔ)上,根據(jù)具體情況靈活運(yùn)用。應(yīng)注意根據(jù)手性藥物的處方與制備拘泥工藝�����、手性中心的多寡與構(gòu)型的穩(wěn)定性�、各立體異構(gòu)體雜質(zhì)生成的可能性與毒性等情況,采用合適的分析方法分別從起始原料、工藝過程與終產(chǎn)品的質(zhì)量標(biāo)準(zhǔn)等各個方面對手性藥物的光學(xué)純度進(jìn)行全程的監(jiān)控,以充分保證手性藥物的質(zhì)量是穩(wěn)定可控的�����。