1����、臨床開發(fā)計劃介紹
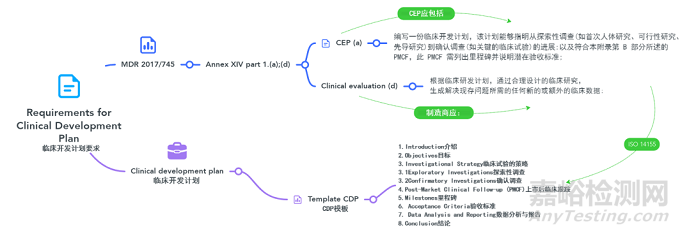
在臨床評估醫(yī)療器械的背景下,制造商必須根據MDR法規(guī)Annex XIV-臨床評價和上市后臨床跟蹤-CLINICAL EVALUATION AND POST-MARKET CLINICAL FOLLOW-UP中概述的規(guī)定�����,在其評估計劃中納入臨床開發(fā)計劃����。
重要的是要注意這些是不同的實體。把這種關系想象成一組嵌套的俄羅斯套娃�����,其中CEP包含了CDP�����。然而在目前的法規(guī)中��,臨床開發(fā)計劃(clinical development plan)僅被提及兩次����,另外請確保要符合ISO 14155。
2、I. General purpose
在本節(jié)中����,制造商將簡要概述正在考慮的醫(yī)療器械及其預期用途。此外�����,制造商將強調制定符合MDR法規(guī)要求的全面臨床開發(fā)計劃的重要性��。
介紹為后續(xù)臨床開發(fā)計劃的制定奠定了基礎�����,展示了其在確保醫(yī)療器械的安全性��、有效性和合規(guī)性方面的重要作用��。
在傳達器械的預期用途時����,強調清晰和精確的重要性��。這一初始背景將為理解臨床開發(fā)計劃的后續(xù)部分奠定基礎����。此外����,強調MDR法規(guī)定義的監(jiān)管環(huán)境�����,這需要創(chuàng)建一個細致而徹底的臨床開發(fā)計劃�����,以滿足最高的評估和批準標準�����。
Tips:使用ISO 14155:2020中概述的要求
3��、I. General purpose
在本節(jié)中��,制造商將簡要概述正在考慮的醫(yī)療器械及其預期用途��。此外����,制造商將強調制定符合MDR法規(guī)要求的全面臨床開發(fā)計劃的重要性。
介紹為后續(xù)臨床開發(fā)計劃的制定奠定了基礎,展示了其在確保醫(yī)療器械的安全性�����、有效性和合規(guī)性方面的重要作用��。
在傳達器械的預期用途時��,強調清晰和精確的重要性����。這一初始背景將為理解臨床開發(fā)計劃的后續(xù)部分奠定基礎。此外��,強調MDR法規(guī)定義的監(jiān)管環(huán)境�����,這需要創(chuàng)建一個細致而徹底的臨床開發(fā)計劃��,以滿足最高的評估和批準標準����。
Tips:使用ISO 14155:2020中概述的要求
4����、II. Objectives
在本節(jié)中��,制造商將闡明臨床開發(fā)計劃的核心目標����。這包括闡明計劃的總體目標����,并概述從初步探索性調查到后期確認階段的順序過渡。制造商的重點將是簡潔地描述每個調查的目的��,并闡明每個步驟如何有助于證實器械的安全性和性能��。
臨床開發(fā)計劃的主要目標是建立一個系統(tǒng)的框架,指導調查從探索性階段確認的發(fā)展階段����。每項調查都是故意設計的,以闡明器械的安全性�����、有效性和整體性能的特定方面�����,確保其無縫整合到臨床實踐中。
Exploratory Investigations/探索性調查:
探索性調查包括首次人體研究��、可行性評估和試點研究等活動��。這些調查的目的是初步了解器械的潛在好處和局限性����。通過將器械置于受控環(huán)境和有限的患者人群中,試圖確定任何潛在的安全問題�����,并為后續(xù)階段完善調查方法��。這些探索性研究對于形成隨后的確證性研究至關重要��,確保所采取的路徑是明智的和戰(zhàn)略性的�����。
Confirmatory Investigations/確證性研究:
隨著計劃的推進����,焦點轉向確證性研究,其中包括關鍵的臨床研究����。這些研究旨在嚴格評估器械的安全性、有效性和在更廣泛的患者范圍內的表現����。目的有兩個:驗證器械的預期用途,并產生強有力的臨床證據來證實其聲明�����。通過采用控制方案��、不同的患者群體和全面的數據收集��,確保器械的安全性和有效性是完善的�����,為監(jiān)管批準和隨后的臨床實施奠定了基礎��。
探索階段提供了一個基本的理解����,塑造了隨后嚴格評估器械屬性的確認階段。我們精心安排了從探索性研究到確證性研究的無縫進展�����,以確保科學合理且具有臨床意義的證據不斷積累����。這一戰(zhàn)略順序旨在建立器械的可信度,促進監(jiān)管機構����、醫(yī)療保健專業(yè)人員和患者之間的信任。
5��、III. Investigational Strategy
在本部分中����,制造商將詳細闡述進行臨床研究的策略方法。這需要有系統(tǒng)地分解調查階段�����,包括探索性和確認性階段��。每個階段的計劃,目標,和方法將被闡述����。
3.1 Exploratory Investigations/探索性調查
在探索性研究領域��,該計劃需要啟動初步研究�����,如首次人體試驗、可行性評估和試點研究�����。這些初步調查是揭示器械潛力��、識別挑戰(zhàn)和完善調查途徑的關鍵墊腳石����。
實例:
First-in-Human Studies/首次人體試驗(FIH): 最初的探索階段將包括首次人體研究,旨在評估器械在有限患者群體中的安全性�����、耐受性和初步療效�����。該研究階段將涉及[預期數量]患者��,重點關注器械的直接影響,并確定任何不可預見的不良事件�����。終點將包括安全性����、基本器械功能和臨床有效性的早期指征。
Feasibility Studies/可行性研究:隨后將進行可行性研究��,包括更廣泛的可控患者隊列�����。主要目的是評估器械在現實臨床環(huán)境中的可行性�����,微調操作方面����,并收集臨床表現的初步數據。本試驗將納入大約[預期數量]的患者����,終點將擴展到易用性��、患者接受度和潛在獲益等因素��。
Pilot Studies/先導性研究:隨后�����,先導性研究將進一步完善研究策略�����。有了[預期數量]的參與者,這些研究將側重于優(yōu)化方案�����、數據收集方法和分析程序��。本文的重點轉向收集更全面的數據��,為隨后的確證性研究設計提供信息����。端點將與關鍵的研究,盡管規(guī)模較小。
3.2 Confirmatory Investigations/確證性調查
進入確認階段后,隨著關鍵臨床研究占據中心地位����,研究性方法將加強。這些研究對于確認器械的安全性�����、有效性和在受控但具有代表性的條件下的整體性能至關重要�����。
實例:
Study Design/研究設計:驗證階段將采用隨機�����、雙盲����、安慰劑對照設計,納入與預期目標人群相似的多樣化患者人群����。這種穩(wěn)健的方法允許對器械的性能進行公正的評估,同時堅持嚴格的科學標準��。這項研究將進行跨多個中心加強普遍性。
Endpoints/終點:主要終點將側重于療效��、安全性和臨床效用的可量化指標�����。這些終點將根據器械的預期用途及其對患者預后的預期影響進行仔細選擇����。次要終點將為患者報告的結果和長期器械性能提供補充見解。
Inclusion/Exclusion Criteria 納入/排除標準:制定明確的納入和排除標準����,以確保納入具有代表性的患者資料,同時排除混雜因素�����。這種方法保證了研究結果反映了現實世界的情況����,并徹底評估了器械的安全性和有效性����。
所選擇的研究策略是由精心設計的計劃所驅動,該計劃將探索性見解與確證性研究的嚴格要求相結合。通過堅持這一戰(zhàn)略藍圖����,我們的目標是產生強有力的臨床證據,證明該器械的安全性�����、有效性�����,并符合MDR法規(guī)的嚴格要求��。
6�����、IV. 4. Post-Market Clinical Follow-up (PMCF)
本節(jié)深入研究執(zhí)行上市后臨床跟蹤(PMCF)研究的綜合計劃�����,如MDR法規(guī) AnnexXIV的Part B所述����。它概述了目標����,方法��,數據收集策略����,以及PMCF在真實臨床環(huán)境中器械性能持續(xù)評估中的關鍵作用。
Explain the Plan/解釋計劃:PMCF研究是整體臨床開發(fā)戰(zhàn)略不可或缺的組成部分��,旨在監(jiān)測器械上市后的實際性能�����。其目的是收集補充臨床數據����,驗證器械的長期安全性和有效性,并識別在上市前評估階段可能不明顯的任何潛在新風險或益處��。
4.1 Objectives/目標
PMCF研究的核心目標包括:
•長期安全性評估:在不同的患者群體中持續(xù)評估器械的安全性����。
•性能驗證:在不同的臨床情況下��,驗證器械的持續(xù)有效性及其對預期臨床目的的依從性。
•識別新出現的問題:檢測任何以前未檢測到的不良事件��、設備相關并發(fā)癥或在實際使用過程中可能出現的益處��。
4.2 Methodologies/方法
PMCF studies將采用多中心�����、前瞻性設計����,以確保結果的穩(wěn)健性和普遍適用性。將模擬真實世界的臨床環(huán)境����,涉及更廣泛和更多樣化的患者人群。這將能夠收集反映器械在實際操作中的性能的代表性數據�����。
•Data Collection Methods/數據收集方法:數據收集將是多方面的����,包括患者報告的結果、臨床評估和設備特定指標的組合��。病人的經歷、治療結果和潛在的并發(fā)癥將被記錄下來����,以評估器械對病人生活質量和整體健康的影響。
•Role of PMCF/PMCF的作用:PMCF在確保器械在初始市場進入后的性能持續(xù)評估方面起著關鍵作用�����。它通過識別增強的領域�����,解決任何不可預見的問題����,并根據實際數據改進器械的功能,從而促進持續(xù)改進��。從PMCF研究中獲得的見解為監(jiān)管機構�����、醫(yī)療保健提供者和患者提供了信息��,有助于提高患者安全性和器械的整體臨床效用�����。
7��、V. Milestones
本節(jié)概述了每個調查階段的關鍵里程碑的時間軸�����,提供了項目進展的結構化概述��。它包括探索性和確證性研究的估計開始和結束日期�����。此外�����,它提出了上市后臨床跟蹤PMCF研究的預期執(zhí)行時間�����。
5.1 Exploratory Investigations/探索性調查
First-in-Human Studies: [Name] [Description]
Estimated Start Date: [Month, Year]
Estimated Completion Date: [Month, Year]
Feasibility Studies: [Name] [Description]
Estimated Start Date: [Month, Year]
Estimated Completion Date: [Month, Year]
Pilot Studies: [Name] [Description]
Estimated Start Date: [Month, Year]
Estimated Completion Date: [Month, Year]
5.2 Confirmatory Investigations/確認性調查
Pivotal Clinical Studies: [Name] [Description]
Estimated Start Date: [Month, Year]
Estimated Completion Date: [Month, Year]
Data Analysis and Interpretation: [Description]
Estimated Start Date: [Month, Year]
Estimated Completion Date: [Month, Year]
Study Planning and Protocol Development: [Description]
Estimated Start Date: [Month, Year]
Estimated Completion Date: [Month, Year]
Recruitment and Data Collection: [Description]
Estimated Start Date: [Month, Year]
Estimated Completion Date: [Month, Year]
Data Analysis and Reporting: [Description]
Estimated Start Date: [Month, Year]
Estimated Completion Date: [Month, Year]
上述里程碑為及時執(zhí)行各研究階段和總體臨床開發(fā)計劃提供了路線圖��。預計時間表允許全面了解項目的持續(xù)時間����,并促進有效的項目管理����。遵守這些里程碑將確保在研究階段的簡化進展�����,最終產生強有力的臨床證據�����,證明器械的安全性��、有效性和遵守MDR法規(guī)中規(guī)定的標準�����。
8�����、VI. Acceptance Criteria
本部分概述了每個研究階段的預先定義的接受標準��,闡明了表明每個研究成功完成和為進入下一階段做好準備的標準。清晰且定義明確的驗收標準可作為基準����,以確保每個研究階段都得到嚴格執(zhí)行��,并與總體臨床開發(fā)計劃保持一致����。
6.1 Exploratory Investigations/探索性調查
1.首次人體試驗FIH研究/First-in-Human Studies
驗收標準:成功完成需要安全數據的收集,潛在不良事件的識別��,以及對器械功能和可行性的初步了解��。本試驗未發(fā)生嚴重不良事件��,且患者耐受性良好�����,這表明患者已為下一階段做好準備�����。
2. 可行性研究/Feasibility Studies
驗收標準:成功完成的標志是操作挑戰(zhàn)的識別����,調查方法的可行性評估��,以及證明器械臨床潛力的初步數據��。收購價值的可行性見解和一個可行的操作策略表示試點研究的準備����。
3.試點研究/Pilot Studies
驗收標準:成功完成包括數據收集方法的改進����,基于試驗數據的方案調整,以及在受控環(huán)境下對器械性能趨勢的理解�����。穩(wěn)健的數據收集過程和有前景的初步研究結果表明����,我們?yōu)榇_證性研究做好了準備。
6.2 Confirmatory Investigations/確認性調查
1. 關鍵臨床研究/Pivotal Clinical Studies
驗收標準:成功完成需要全面的數據收集��、統(tǒng)計分析和生成強有力的證據來支持器械的安全性和有效性����。對于主要終點�����,達到預先設定的統(tǒng)計學顯著性閾值并證明患者結局良好將意味著該階段的成功實施�����。
2. 數據分析與解讀/
Data Analysis and Interpretation
驗收標準:成功完成包括徹底的數據分析,主要和次要終點的統(tǒng)計驗證�����,以及與器械預期用途相一致的臨床證據的綜合�����。如果結果的解讀符合預先設定的成功標準和目標�����,將表明準備好參加PMCF研究��。
6.3 Post-Market Clinical Follow-up (PMCF) Study
1. 研究計劃和方案制定/
Study Planning and Protocol Development
接受標準:成功完成包括制定全面的PMCF研究方案����、確定適當的終點和明確的患者人群��。研究方案與監(jiān)管要求的一致性以及利益相關方的批準為繼續(xù)執(zhí)行研究奠定了基礎��。
2. 招募和數據收集
接受標準:成功完成試驗包括招募足夠的患者隊列����、無縫收集數據以及遵循既定方案�����。達到所需的樣本量并獲取與研究方案一致的相關臨床數據將表明為最終階段做好準備��。
3.數據分析與報告
驗收標準:成功完成包括全面的數據分析�����、研究結果的綜合和結果的報告��。提供關于器械長期安全性和有效性的結論性證據����,加上遵守監(jiān)管報告要求,標志著PMCF研究目標的實現����。
所描述的驗收標準為每個調查階段的成功執(zhí)行和到后續(xù)階段的過渡提供了一個清晰的路線圖����。這些標準與監(jiān)管標準和臨床目標保持一致�����,確保每個階段的目標都得到滿足��,并且器械通過臨床開發(fā)計劃的旅程是系統(tǒng)和全面的��。
9��、VII. Data Analysis and Reporting
在本部分中�����,制造商將提出一份全面的數據分析計劃����,包括統(tǒng)計學方法��、終點評估和安全性評估��。該部分將闡明如何根據相關法規(guī)和指南進行數據收集�����、分析和報告,確保對整個研究階段產生的臨床證據進行嚴格審查��。
7.1 數據分析策略/Data Analysis Strategy
數據分析計劃將涉及多方面的方法��,包括嚴格的統(tǒng)計學方法��、終點評估和安全性評估��。統(tǒng)計分析將用于量化器械的有效性和安全性結果��,同時也闡明收集數據中的模式��。將根據數據類型和研究設計����,并遵循既定的最佳實踐,選擇嚴格的統(tǒng)計方法����。
7.2 終點評價/Endpoint Evaluation
我們將仔細評估主要和次要終點。與器械預期臨床獲益直接相關的主要終點����,將評估其統(tǒng)計學意義和臨床相關性�����。次要端點��,提供補充的見解�����,將被分析以增強對器械影響的整體理解��。
7.3 安全性評估/Safety Assessments
安全性評估將是數據分析的重要組成部分����。不良事件����、并發(fā)癥和安全相關趨勢將被徹底分析�����,以評估器械的安全性��。該評估將涉及對不同患者群體�����、用藥持續(xù)時間和臨床場景的安全性數據進行系統(tǒng)檢查。
7.4 數據收集�����、分析和報告過程
Data Collection, Analysis, and Reporting Process
數據收集將遵循嚴格的方案����,以確保一致性和準確性。在分析之前�����,收集的數據將經過徹底的驗證和驗證過程����。我們將使用統(tǒng)計學工具評估臨床結局的顯著性,而遵循預先設定的統(tǒng)計學顯著性水平將指導結果的解讀�����。
報告過程將包括創(chuàng)建符合適用法規(guī)和準則的全面報告����。研究結果將透明地提交�����,展示臨床證據的優(yōu)勢和局限性����。此外��,數據報告的結構將有助于與監(jiān)管機構�����、醫(yī)療專業(yè)人員和其他利益攸關方進行明確溝通����。
7.5 法規(guī)遵從性和指引/Regulatory Compliance and Guidelines
所有數據分析和報告活動將嚴格遵守MDR法規(guī) (MDR )和其他相關指南中概述的監(jiān)管要求。統(tǒng)計學方法的選擇��、結局的解釋和結果的呈現將以這些法規(guī)為指導��,以確保穩(wěn)健性����、透明度和法規(guī)合規(guī)性��。
10、VIII. Conclusion
本結論部分概括了臨床開發(fā)計劃的關鍵方面����,強調了其綜合本質及其在保證器械的安全性和性能方面的關鍵作用,符合MDR法規(guī)的規(guī)定����。
8.1 本計劃的全面性質/
Comprehensive Nature of the Plan
臨床開發(fā)計劃作為精心制作的藍圖,指導器械通過一系列明確定義的研究階段����。通過將探索性調查納入確證性研究,并在上市后臨床跟蹤(PMCF)中達到高潮�����,該計劃提供了一種全面的方法來生成可靠的臨床證據����,以證實器械的預期用途、安全性和有效性��。
8.2 在保證器械安全和性能中的作用
臨床開發(fā)計劃的核心是承諾確?����;颊甙踩推餍敌阅堋T撚媱澋墓δ苁欠婪稘撛诘娘L險和不確定性��,編排一個戰(zhàn)略途徑����,系統(tǒng)地評估器械在不同患者情況下的屬性。該計劃是創(chuàng)新和患者健康之間的橋梁�����,使我們能夠彌合理論潛力和真實世界療效之間的差距����。
8.3 符合MDR 要求
臨床開發(fā)計劃的制定是由堅定不移地遵守MDR法規(guī)中概述的要求驅動的。通過嚴格遵守這些規(guī)定��,該計劃確保從探索性評估到確證性研究和PMCF的每個調查階段都符合最高的監(jiān)管標準����。這種一致性不僅對于法規(guī)遵從性至關重要,而且對于在監(jiān)管機構�����、醫(yī)療專業(yè)人員和患者等之間灌輸信心至關重要��。
總之�����,臨床開發(fā)計劃不僅僅是一種程序義務;這是一項戰(zhàn)略要求����,支撐著器械從概念到臨床現實的旅程。通過精心制定這一計劃����,我們?yōu)閲栏裨u估、證實證據以及確?����;颊甙踩透l淼於嘶A��。通過其全面的性質和與MDR 要求的一致性����,臨床開發(fā)計劃體現了我們對開拓醫(yī)療創(chuàng)新的承諾,同時堅持最高的法規(guī)遵從和患者護理標準�����。