A:制劑所含有的雜質(zhì)種類都有什么�?
B:制劑中的雜質(zhì)統(tǒng)稱為降解產(chǎn)物,包括原料藥的降解產(chǎn)物�、原料藥與賦形劑或包裝容器的反應(yīng)產(chǎn)物。不包括存在于新原料藥中的雜質(zhì)���,除非它們也屬于降解產(chǎn)物���;不包括從新藥制劑的賦形劑或從包裝容器滲出產(chǎn)生的雜質(zhì);不包括生物及生物制品�、縮肽、寡聚核苷酸�、放射性藥物、發(fā)酵制品及其半合成品����、草藥和來源于動植物的粗制品�;另外也不包括外源性污染物����、多晶型和對映體雜質(zhì)。
A:雜質(zhì)研究是藥品質(zhì)量研究的重要內(nèi)容�,在藥品研發(fā)的過程中,如何合理地報(bào)告和控制降解產(chǎn)物�?
B:關(guān)于降解產(chǎn)物報(bào)告和控制的合理性部分,有以下幾個(gè)要點(diǎn):申報(bào)者應(yīng)對新藥制劑的生產(chǎn)和(或)穩(wěn)定性考察中所發(fā)現(xiàn)的降解產(chǎn)物進(jìn)行綜述����。該綜述應(yīng)包括對制劑中可能的降解途徑和因與賦形劑和(或)包裝容器反應(yīng)所產(chǎn)生雜質(zhì)的科學(xué)評價(jià)。對新藥制劑降解產(chǎn)物檢測的所有實(shí)驗(yàn)室研究工作進(jìn)行總結(jié)����,包括研發(fā)過程中生產(chǎn)的批次和采用擬上市工藝生產(chǎn)批次的試驗(yàn)結(jié)果,并對比和討論這兩者批次的雜質(zhì)概況����,存在的任何差異都應(yīng)進(jìn)行討論。應(yīng)對不屬于降解產(chǎn)物的雜質(zhì)(如來自于原料藥的工藝雜質(zhì)和由賦形劑產(chǎn)生的雜質(zhì))進(jìn)行說明�。穩(wěn)定性考察中發(fā)現(xiàn)的任何降解產(chǎn)物,如果大于鑒定限度時(shí)���,則應(yīng)對其作結(jié)構(gòu)確證�。若無法確證某一降解產(chǎn)物結(jié)構(gòu)時(shí),也應(yīng)在申報(bào)資料中說明實(shí)驗(yàn)室對鑒定該物質(zhì)已作過的所有研究工作���。對不大于限度的降解產(chǎn)物通常不需要確證其結(jié)構(gòu)���。但是,對于那些可能有不尋常功效或產(chǎn)生毒性藥理作用的降解產(chǎn)物���,即使不大于鑒定限度���,仍應(yīng)建立分析方法����。
A:對于各批次產(chǎn)品降解產(chǎn)物報(bào)告,在內(nèi)容上有什么具體要求����?
B:這部分的要求主要有以下幾點(diǎn):申報(bào)資料中應(yīng)提交用于臨床、安全性���、穩(wěn)定性試驗(yàn)的所有相關(guān)批次以及采用擬上市工藝生產(chǎn)的有代表性批次的新藥制劑的分析結(jié)果���;定量測定結(jié)果應(yīng)數(shù)字化�,不應(yīng)使用類似“符合規(guī)定”“符合限度”等一般性術(shù)語���;應(yīng)報(bào)告新藥制劑相關(guān)批次中檢測到的任何大于報(bào)告限度的降解產(chǎn)物以及總的降解產(chǎn)物�,并附所用分析方法����;d若降解產(chǎn)物含量小于1.0%時(shí),結(jié)果報(bào)告至小數(shù)點(diǎn)后兩位(如0.06%)���;大于等于1.0%時(shí)�,應(yīng)報(bào)告至小數(shù)點(diǎn)后一位(如:1.3%)���;降解產(chǎn)物應(yīng)用編號或適當(dāng)?shù)拿枋霰硎荆ㄈ绫A魰r(shí)間“雜質(zhì)RRT=0.37”)�;所有大于報(bào)告限度的降解產(chǎn)物都應(yīng)進(jìn)行累加并以“總雜質(zhì)”報(bào)告�;申報(bào)資料中應(yīng)提供代表性樣品批次的有標(biāo)記峰的色譜圖(或采用其他方法獲得的相關(guān)數(shù)據(jù)),包括在分析方法驗(yàn)證中和長期�、加速穩(wěn)定性研究中所得到的色譜圖。申報(bào)者應(yīng)能確保�,如管理部門需要,可提供每個(gè)批次的完整降解產(chǎn)物概況(如色譜圖)�;申報(bào)資料中應(yīng)提供每一批次新藥制劑的詳細(xì)信息����,具體內(nèi)容如下:批號���、規(guī)格和批量���、生產(chǎn)日期、生產(chǎn)地點(diǎn)���、生產(chǎn)工藝���、直接接觸的包裝容器、降解產(chǎn)物的含量����,單個(gè)的和總量�、批次的用途(如臨床研究、穩(wěn)定性研究)���、分析方法所用的對照品����、用于該制劑的原料批號、穩(wěn)定性研究的放置條件���。
A:對于降解產(chǎn)物的分析方法����,有沒有方法學(xué)驗(yàn)證的要求或者其他要求���?
B:首先分析方法應(yīng)按照ICHQ2進(jìn)行驗(yàn)證并適用于降解產(chǎn)物的定性和定量檢測�,尤為重要的是�,應(yīng)能證明分析方法具有檢測特定或非特定降解產(chǎn)物的專屬性。必要時(shí)����,還應(yīng)包括對放置在相對強(qiáng)烈條件(光、熱�、濕、酸/堿和氧化)下的樣品所進(jìn)行的分析方法驗(yàn)證���。當(dāng)分析方法揭示除降解產(chǎn)物以外還存在其他色譜峰(如原料藥�,原料藥合成時(shí)引入的雜質(zhì)���,賦形劑和由賦形劑產(chǎn)生的雜質(zhì))�,這些峰需在色譜圖中進(jìn)行標(biāo)注,在驗(yàn)證文件中應(yīng)探討他們的來源�;其次,分析方法的定量限應(yīng)不大于報(bào)告限度���;最后����,降解產(chǎn)物的量可以用一系列的技術(shù)手段測定(如內(nèi)標(biāo)法�、主成分自身對照法、加校正因子的主成分自身對照法等)����。
A:新藥制劑的質(zhì)量標(biāo)準(zhǔn)中,對于降解產(chǎn)物的檢查項(xiàng)目有什么要求�?
B:新藥制劑的質(zhì)量標(biāo)準(zhǔn)中應(yīng)包括在上市產(chǎn)品生產(chǎn)和推薦的貯藏條件下預(yù)期會出現(xiàn)的降解產(chǎn)物檢查項(xiàng)目。穩(wěn)定性研究����、降解途徑的了解、產(chǎn)品開發(fā)研究以及實(shí)驗(yàn)室研究都可用來確定降解的概況����。有以下幾點(diǎn)要求:新藥制劑質(zhì)量標(biāo)準(zhǔn)中列入的降解產(chǎn)物檢查項(xiàng)����,應(yīng)根據(jù)擬上市工藝生產(chǎn)的批次中發(fā)現(xiàn)的降解產(chǎn)物來確定����。應(yīng)對在安全性研究���、臨床研究和穩(wěn)定性研究中觀察到的降解產(chǎn)物狀況�、并結(jié)合擬上市工藝生產(chǎn)的產(chǎn)品批次中降解產(chǎn)物的狀況兩者綜合進(jìn)行討論�,再對質(zhì)量標(biāo)準(zhǔn)中列入和不列入哪些降解產(chǎn)物的理由進(jìn)行說明;指導(dǎo)原則中有引入一個(gè)“特定降解產(chǎn)物”概念�,特定降解產(chǎn)物是指列入新藥制劑質(zhì)量標(biāo)準(zhǔn)中有特定限度要求的各個(gè)降解產(chǎn)物。特定降解產(chǎn)物可以是結(jié)構(gòu)已確證和未確證的�;質(zhì)量標(biāo)準(zhǔn)中應(yīng)包含特定的已鑒定的降解產(chǎn)物和估計(jì)含量大于鑒定限度的特定結(jié)構(gòu)未鑒定降解產(chǎn)物;特定的未鑒定降解產(chǎn)物應(yīng)用適當(dāng)?shù)姆椒▉順?biāo)示�,如:“未鑒定雜質(zhì)A”“相對保留時(shí)間為0.9的雜質(zhì)”;對于被認(rèn)為具有特殊功能或產(chǎn)生藥理毒性或未預(yù)料到的藥理作用的降解產(chǎn)物���,其分析方法的定量限/檢測限必須與降解產(chǎn)物被控制的量相當(dāng)���;對于任何一個(gè)非特定的降解產(chǎn)物應(yīng)有一個(gè)不大于鑒定限度的認(rèn)可標(biāo)準(zhǔn),對總降解產(chǎn)物也應(yīng)建立一個(gè)認(rèn)可標(biāo)準(zhǔn)���;對于指定的降解產(chǎn)物�,制訂其認(rèn)可標(biāo)準(zhǔn)時(shí),應(yīng)考慮其在原料藥中的認(rèn)可標(biāo)準(zhǔn)(如有的話)���,它的通常含量以及它在穩(wěn)定性研究中建議的有效期和推薦的貯存條件下的增加量����。當(dāng)然���,認(rèn)可標(biāo)準(zhǔn)的設(shè)定不得高于該降解產(chǎn)物經(jīng)界定的安全含量���。簡要概括,新藥制劑質(zhì)量標(biāo)準(zhǔn)中應(yīng)包括以下降解產(chǎn)物的檢查項(xiàng):每種特定的�、已鑒定降解產(chǎn)物;每種特定的未鑒定降解產(chǎn)物���;任何不大于鑒定限度認(rèn)可標(biāo)準(zhǔn)的非特定降解產(chǎn)物���;降解產(chǎn)物總量。
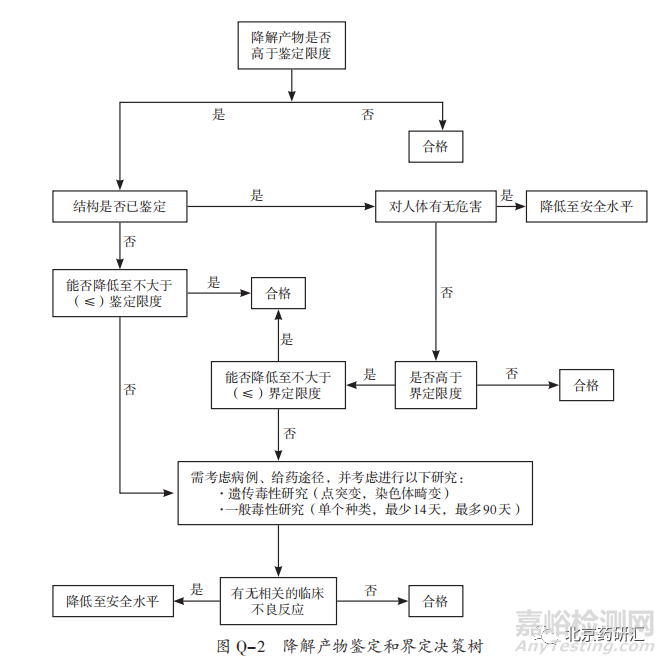
A:降解產(chǎn)物的界定是什么意思�?
B:首先引入雜質(zhì)的界定概念。雜質(zhì)的界定是獲得和評價(jià)某些數(shù)據(jù)的過程���,這些數(shù)據(jù)可用于確保單個(gè)雜質(zhì)或在特定含量下的一系列雜質(zhì)的生物安全性����。對于一個(gè)通過充分的安全性研究和臨床研究的新藥制劑���,其中任何一個(gè)降解產(chǎn)物的水平即被認(rèn)為是已經(jīng)通過了界定的�。對于是動物和(或)人體中重要代謝物的降解產(chǎn)物���,通常也視為已通過界定����。如果建立的認(rèn)可標(biāo)準(zhǔn)超過界定限度���,而所獲得的試驗(yàn)數(shù)據(jù)不能用來證明降解產(chǎn)物的認(rèn)可標(biāo)準(zhǔn)限度是合理的�,則必須進(jìn)行進(jìn)一步研究���。也可以利用降解產(chǎn)物鑒定和界定的決策樹來進(jìn)行判斷�,這個(gè)決策樹在上個(gè)章節(jié)Q3A新型原料藥中的雜質(zhì)問題中已有講解���。