在質(zhì)量管理中,偏差是質(zhì)量保證要素的關鍵部分——本文從藥品研發(fā)實施偏差管理的意義���、研發(fā)偏差的特點�����、實施流程���、常見問題等方面進行闡述,圍繞藥品研發(fā)工作的特點和目的提出了對研發(fā)實施偏差的見解���,以期為更多藥品研發(fā)人員提供思路�,增加制藥業(yè)同仁們對偏差的認識�����,為建立適合研發(fā)的偏差管理程序���、在日常工作中正確地開展偏差調(diào)查�����、研發(fā)過程中質(zhì)量體系的建立與完善提供助力�����。
近幾年�,制藥行業(yè)在藥品注冊、法規(guī)�����、監(jiān)管方面發(fā)生了一系列重大事件:2015年���,國家食品藥品監(jiān)督管理總局開展臨床試驗數(shù)據(jù)核查�,發(fā)現(xiàn)業(yè)內(nèi)存在研發(fā)試驗數(shù)據(jù)不可靠�����、研發(fā)過程缺少合規(guī)管理等現(xiàn)象�����;2017年�����,我國加入ICH(國際人用藥品技術要求協(xié)調(diào)會),國內(nèi)研發(fā)進一步受到ICH Q8、Q9���、Q10的影響,QbD(質(zhì)量源于設計)理念更加深入;2019年���,新修訂的《中華人民共和國藥品管理法》出臺,提出落實“四個最嚴”精神……這些事件讓越來越多的藥品研發(fā)機構或藥品生產(chǎn)企業(yè)的研發(fā)部門開始正視研發(fā)環(huán)節(jié)的質(zhì)量管理[1-4]���。
在質(zhì)量管理中�,偏差是質(zhì)量保證要素的關鍵部分�����,偏差調(diào)查可以直接體現(xiàn)出一個公司質(zhì)量管理的整體情況,也是公司實現(xiàn)質(zhì)量持續(xù)改進的關鍵環(huán)節(jié)[5]�����?����;诖?��,偏差管理也始終是監(jiān)管部門檢查的重點。
國內(nèi)藥品監(jiān)管的“偏差”一詞來源于《藥品生產(chǎn)質(zhì)量管理規(guī)范(2010年修訂版)》(即GMP)�,其應用主要在藥品生產(chǎn)過程控制環(huán)節(jié)[6]���,常見的審計缺陷案例也主要為藥品生產(chǎn)過程中的偏差事例�����,所以很多研發(fā)人員甚至部分質(zhì)量管理人員都認為偏差只適用于藥品生產(chǎn)過程,而忽略了偏差管理在研發(fā)過程中的重要性�����。
正是因為對偏差的認識不足�����,目前研發(fā)機構的偏差管理非常混亂�,有些機構缺少偏差管理程序�����,即使有偏差產(chǎn)生也沒有認識到�;有些機構雖然起草了偏差管理的SOP(標準作業(yè)程序)�,但是在執(zhí)行層面上仍然對偏差處理存在困惑�。這些問題導致藥品研發(fā)機構的偏差管理既不能滿足法規(guī)監(jiān)管的要求,也無法發(fā)揮偏差作為質(zhì)量管理體系改進工具的作用�。對此�����,本文接下來將對偏差管理的特點�、實施流程及常見問題等進行探討�,為藥品研發(fā)人員提供思路�����。
1�、偏差管理的意義
1.1提高人員技術水平、增加技術和經(jīng)驗積累
偏差的識別���、調(diào)查、糾正及整改預防措施的制定�,每一個環(huán)節(jié)都需要參與的人員有足夠的經(jīng)驗和技術能力�����,這在無形中能增加個人及團隊經(jīng)驗技術的知識積累�。
1.2改變和提升管理理念
ICH Q10中的“知識管理”理念強調(diào)對產(chǎn)品和工藝知識進行管理應從研發(fā)階段開始���。運用科學方法開展研發(fā)活動即是為產(chǎn)品和工藝理解提供知識[7]。偏差作為一種發(fā)現(xiàn)問題�、分析問題�、解決問題并跟蹤改進直到問題解決至可接受的風險水平的有效手段�,能促使管理人員形成系統(tǒng)化的產(chǎn)品和工藝知識體系,對工藝性能和產(chǎn)品質(zhì)量的持續(xù)改進具有重要意義�����。
1.3標本兼治�、降低成本
偏差調(diào)查是查找根本原因并徹底糾正的過程���,在調(diào)查時不允許淺嘗輒止,糾正時不能“頭痛醫(yī)頭腳痛醫(yī)腳”�����,這種以終為始的工作思路能夠有效地防止類似問題不再發(fā)生�,顯著降低企業(yè)成本�����。
2、研發(fā)偏差的特點
2.1定義
2.1.1?法規(guī)的定義
ICH Q7對“偏差”的定義為:偏離批準的指南或制定的標準[8]���。ICH Q7這里的“標準”,可理解為為實現(xiàn)藥品質(zhì)量而建立的各種技術標準�����,包括但不僅限于產(chǎn)品的質(zhì)量標準�,也可以指其他技術標準,如倉儲物料管理程序�、實驗室程序�����、數(shù)據(jù)管理等���。GMP在第十章第五節(jié)里規(guī)定了偏差處理的要求,以舉例形式細化了偏離的具體情形���,但并沒有給出“偏差”的正式定義[9]。
2.1.2?研發(fā)偏差的定義
研發(fā)機構在解讀和定義研發(fā)偏差時應結合研發(fā)固有的特定目的�����,即短期為研究工藝���、制定產(chǎn)品質(zhì)量標準,長遠為保障公眾用藥�����、促進公眾健康�����,對“偏離批準的指南或制定的標準”的情況進行具體細化���,體現(xiàn)出與藥品生產(chǎn)偏差的差異�����。故研發(fā)偏差可以定義為:對所研究的實驗結果及結論���、被研究產(chǎn)品的關鍵工藝參數(shù)(CPP)和關鍵質(zhì)量屬性(CQA)、質(zhì)量管理系統(tǒng)及程序�����、產(chǎn)品質(zhì)量存在影響或潛在影響的偏離行為���。
2.2適用范圍
工藝研究、小試�、中試、BE(生物等效性)�、臨床試驗用樣品制備���、工藝驗證各個階段,都存在影響或潛在影響研發(fā)實驗結果及結論的行為���,這些行為均應通過標準管理程序進行規(guī)定�����,如組織機構與人員、文件與記錄���、設施與設備、物料與樣品���、儀器與設備的確認���、分析方法驗證、實驗室管理�����、數(shù)據(jù)管理�、質(zhì)量保證各要素�、穩(wěn)定性考察等�。只有預先定義了這些規(guī)則�����,偏差管理才能在此基礎上發(fā)揮有效性���。
研發(fā)中常見的偏差行為有:
(1)沒有執(zhí)行已制定的管理程序���。例如�����,沒有按照儀器設備操作規(guī)程進行設備操作�����,儀器設備未按照管理程序進行確認���、校準�、定期維護保養(yǎng)�,造成設備儀器故障或?qū)嶒炇 ⒔Y果不可靠�。
(2)研發(fā)物料在管理過程中���,沒有按照物料儲存條件存放或倉庫環(huán)境超出設定的溫濕度條件�����,可能會造成物料質(zhì)量發(fā)生變化�����;在出入庫物料的過程中出現(xiàn)了混淆�����、差錯等現(xiàn)象�。
(3)分析檢驗過程中發(fā)生的操作偏離�����。例如�,實驗人員未按規(guī)定的要求書寫實驗記錄�,出現(xiàn)數(shù)據(jù)不可靠的風險���;新人入職后未按培訓管理的要求先進行理論和技術培訓�,在不確定是否滿足上崗的情況下直接安排從事實驗操作�����,實驗失敗或?qū)嶒灲Y果不可靠的風險極大���;樣品及試劑試液的使用�、配制���、有效期錯誤;實驗室的溫濕度���、環(huán)境光照等不滿足檢測要求等。
2.3人員職責和資質(zhì)
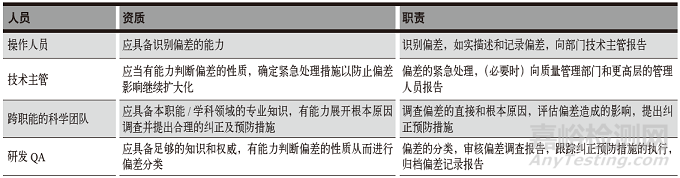
在研發(fā)質(zhì)量管理體系中往往存在質(zhì)量職能不完善�����,QA(質(zhì)量保證)兼職多項工作的情況�����,而偏差調(diào)查的目的和程序決定了其不是一個人或一個部門的事情�����,復雜的調(diào)查甚至涉及多個跨職能部門���。明確相關人員的職責是順利進行偏差調(diào)查、找出根本原因并進行糾正的關鍵���。在研發(fā)偏差管理中���,對相關人員資質(zhì)及職責的要求可參考表1�����。
表1 人員資質(zhì)及職責要求
3�、研發(fā)偏差實施流程和關鍵控制點
3.1實施流程
3.1.1?偏差處理的基本流程
偏差處理流程為:偏差發(fā)現(xiàn)→緊急控制→調(diào)查(書面報告整理)→確定根本原因/直接原因→影響評估→制定糾正及預防措施→跟蹤和評價→關閉批準→歸檔[10-11]�。在偏差處理邏輯上�,研發(fā)與生產(chǎn)沒有太大區(qū)別���,但是在每個環(huán)節(jié)的具體規(guī)定和操作上有研發(fā)專屬性的差異。
研發(fā)的工作性質(zhì)決定了其存在大量的不確定性和不可預見性���,這也是研發(fā)與商業(yè)化生產(chǎn)最大的區(qū)別。研發(fā)在制定偏差管理程序時應向GMP靠攏�,但又不能完全照搬照套商業(yè)化生產(chǎn)的偏差程序。在技術創(chuàng)新和規(guī)范操作之間進行協(xié)調(diào)和平衡是建立研發(fā)質(zhì)量管理程序的立足根本�。
研發(fā)進行質(zhì)量管理時�����,在不同階段應遵守不同的要求�����,防止過度與不足�����。過度會束縛研發(fā)的創(chuàng)新和項目研究進度,不足會出現(xiàn)質(zhì)量管理失控與數(shù)據(jù)不可靠的問題�����。所以在設計研發(fā)偏差管理程序時應基于藥品使用風險和企業(yè)資源�����、研究成本等進行綜合考慮�����,GMP臨床試驗用藥品附錄中就強調(diào)了臨床試驗用藥品應當基于風險建立質(zhì)量管理體系[12]���。
3.1.2?研發(fā)偏差分類
研發(fā)階段的偏差可以按兩種形式進行:簡易偏差和完整偏差調(diào)查�����。例如���,對于早期創(chuàng)新藥先導化合物篩選�、仿制藥工藝處方篩選���、分析方法開發(fā)階段出現(xiàn)的偏離行為應進行簡易偏差記錄,此時應降低調(diào)查力度�����,重點關注操作的規(guī)范性�����;對于此類簡易偏差在處理過程中可以弱化關閉控制、簡化調(diào)查記錄���,批準的權利可下放至實驗室技術主管而非必須由質(zhì)量管理部門把控。在中后期工藝優(yōu)化階段應進行完整偏差調(diào)查�����,如對于臨床試驗用藥品應當對偏離制備工藝�����、質(zhì)量標準的情況以及其他可能影響臨床試驗用藥品質(zhì)量的偏差進行詳細的調(diào)查評估���。
3.2關鍵控制點
3.2.1?偏差的識別
這是偏差處理活動的起點���。前面所述的對偏差進行清晰明確的定義是識別偏差的基礎。另外必須對一線實驗員進行偏差知識的培訓���,確保人員在具備崗位工作經(jīng)驗和技術能力的基礎上樹立正確的偏差意識�����。
3.2.2?發(fā)動及報告時限
對于偏差的上報�����,GMP要求立即報告本部門主管人員和QA,但是卻沒有對“立即”給出明確的定義���,研發(fā)機構在起草偏差處理程序時,應結合本公司實際情況在規(guī)程中給出“立即”的定義�,以明確指導員工。例如�,偏差發(fā)現(xiàn)后半個工作日之內(nèi)進行報告���,如果晚上加班無法直接上報,可延期至第二天�����。
立即上報主管是為了能在第一時間內(nèi)采取有效的緊急控制措施,減少偏差危害進一步擴大化�����。另外,立即上報是報給本崗位或部門的技術主管���,此時并不強制必須報告給質(zhì)量管理部門。
3.2.3?原因調(diào)查及工具的應用
偏差原因的調(diào)查需要發(fā)生部門�����、質(zhì)量管理部�、其他相關部門(如倉儲���、工程設備等部門)共同參與,是個團隊合作工作�,絕對不能由偏差發(fā)生部門尤其是偏差發(fā)生人員自己完成調(diào)查。對于原因顯而易見的錯誤�����,可直接通過更正就地解決���;而對于重大偏差或關鍵偏差,通常需要更多全面客觀的深入調(diào)查來挖掘根本原因���。也只有進行全面系統(tǒng)的調(diào)查才能找到真正的根本原因���,才能進行有效的糾正及預防�。
調(diào)查過程應緊密圍繞五大關鍵要素“人���、機���、料�、法、環(huán)”���,采取失敗模式(FIMA)、頭腦風暴法�����、魚骨圖分析���、5WHY等工具進行逐一排查[13-16]���。
3.2.4?偏差的結論
應根據(jù)“初始偏差信息”���、“進一步調(diào)查”的結果進行綜合的判斷,并對此偏差作出評價:(1)是否對本次實驗結果造成影響�����;(2)是否對本產(chǎn)品的研究結論造成影響�,比如分析檢驗數(shù)據(jù)出現(xiàn)偏差是否影響到了關鍵工藝參數(shù)或關鍵質(zhì)量屬性的判定;(3)是否對注冊造成影響���;(4)是否會對最終生產(chǎn)線上的產(chǎn)品質(zhì)量造成影響�。
3.2.5?糾正和預防措施
在GMP中�����,“偏差管理”與“糾正措施和預防措施(CAPA)”是獨立的兩個章節(jié),但是它們之間有遞進關系���。糾正是基于當下錯誤�����,對當下問題本身進行整改�;而預防措施重點在于防止偏差今后再次發(fā)生���。研發(fā)機構執(zhí)行偏差程序時容易忽略預防,習慣于僅針對偏差事件本身進行糾正�����,這是違背偏差標本兼治理念的�����。另外,在制定預防措施時應客觀全面考慮管理中存在的漏洞和缺陷�,更多地從優(yōu)化或改變流程入手,而不是進行一味的經(jīng)濟處罰�。
3.2.6?偏差的定期回顧
應根據(jù)偏差種類或根源對偏差進行年度趨勢分析。偏差的趨勢分析是管理層定期回顧質(zhì)量體系的一項重要內(nèi)容���,當發(fā)現(xiàn)形成某種趨勢時�,應采取相應的改進行動�����。這也是研發(fā)質(zhì)量管理中容易忽略的一個工作�����,尤其是當研發(fā)質(zhì)量管理崗位非QA出身�,對GMP中質(zhì)量保證要素并不熟悉時。
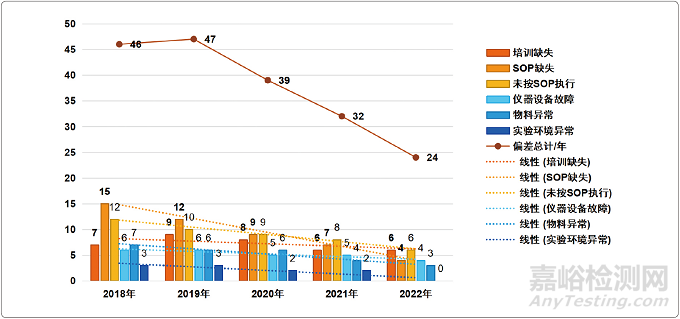
在進行偏差回顧時可以應用統(tǒng)計學知識�����,從不同的角度分析偏差發(fā)生的頻率�����、多發(fā)人員�、多發(fā)部門或?qū)嶒?��,客觀地歸納和總結出偏差管理存在的問題,發(fā)現(xiàn)質(zhì)量管理中的薄弱環(huán)節(jié)�����,從而有的放矢���、提高具體某個環(huán)節(jié)的質(zhì)量管理�����。偏差回顧統(tǒng)計分析案例詳見圖1�。
圖1 某研發(fā)系統(tǒng)趨勢分析統(tǒng)計
案例分析:
(1)從圖1可以直觀看出�����,6大偏差分類項中每一項的數(shù)量均呈逐年遞降的趨勢�,說明該研發(fā)系統(tǒng)采用偏差管理工具后�����,在問題分析及解決方面取得了實效�����,以致于“異常”(偏差)越來越少���。
(2)就單一偏差項分析���,從趨勢線上可以看到���,6大偏差分類中“SOP缺失”的斜率最大���,說明其降幅最大。
(3)從柱狀圖可看到�,6大偏差分類中占比最高的為“SOP缺失”。說明影響質(zhì)量管理的最大因素為SOP(流程制度)的缺失或不完善�����,此為薄弱環(huán)節(jié)�。
綜合(2)、(3)項���,也可以看出該研發(fā)系統(tǒng)在質(zhì)量管理過程中抓住了“SOP缺失”這一要因,在進行偏差糾正和預防措施時���,重點加強了SOP制度流程建設。
(4)趨勢線中���,排名斜率第二的為“實驗室環(huán)境異常”這一偏差項,該項自2018年的3例降至2022年的0例�����。說明實驗室環(huán)境的整改相對容易���。而斜率最低的項為“儀器設備故障”���,雖然每年例次占比不高,但是每年的整改效果并不明顯�����。這為今后質(zhì)量管理提供了方向�����,下一步的改進重點應該放在儀器設備故障���、維護保養(yǎng)等方面�。
(5)最后���,折線圖直觀呈現(xiàn)了該研發(fā)系統(tǒng)自2018年至2022年每年的偏差數(shù)量逐年降低���,說明該研發(fā)系統(tǒng)質(zhì)量管理能力及管理效果逐年提升���。
4、偏差常見問題及原因分析
偏差管理程序主要包括兩大結構:實施和調(diào)查報告的整理���。下面將分別針對兩大結構的常見問題進行總結分析。
4.1實施過程中存在的問題及分析
4.1.1?不上報或立即上報但并不處理
有一些實驗員在知道出現(xiàn)了偏差的情況下�����,害怕調(diào)查帶來的實驗效率降低或懲罰措施而選擇悄悄處理掉錯誤不上報���;也有的實驗員不管出現(xiàn)何種錯誤,全部立即上報給QA�����,讓QA人員全面負責�,實驗室內(nèi)部缺乏調(diào)查的主動性和積極性���。
這兩種做法全都違背了偏差管理的初衷�����。偏差調(diào)查程序存在的目的是為了發(fā)現(xiàn)原因并整改,以更好地維護好質(zhì)量管理體系�,不能只流于形式。出現(xiàn)隱瞞不報或上報后不處理的情況�,關鍵在于人員缺少對偏差理念的正確認識。
4.1.2?調(diào)查人員缺少足夠的知識和經(jīng)驗
在有些企業(yè)的偏差管理程序中�,把偏差調(diào)查僅列為了初級人員的職責范圍而沒有規(guī)定部門主管也有調(diào)查職責�。于是當發(fā)生偏差時,僅有初級人員進行調(diào)查�,他們受經(jīng)驗�����、知識的限制�,并且缺少對偏差宏觀的認識和培訓�,往往缺少刨根問底的動力去深入調(diào)查���,也就不可能實現(xiàn)偏差調(diào)查的初衷[17]。
4.1.3?調(diào)查不夠深入
主要表現(xiàn)為:(1)完成調(diào)查的態(tài)度比較敷衍���;(2)調(diào)查前就已經(jīng)有或者很快就有了結論���;(3)對問題的原因想當然�,不按流程逐項進行調(diào)查���;(4)調(diào)查沒有在發(fā)現(xiàn)后第一時間進行,導致缺少了現(xiàn)場和一些數(shù)據(jù)的丟失�,沒有條件再深入進行�。
出現(xiàn)這些現(xiàn)象的一個重要原因是管理層未能提供足夠的時間�、資源支持來保證調(diào)查的順利進行,調(diào)查人員們沒有足夠的時間和權限進入他們想調(diào)查的區(qū)域���。研發(fā)機構不應只關注項目進度,而不關心項目質(zhì)量�����。
4.1.4?不按SOP進行調(diào)查
主要表現(xiàn)為:公司沒有制定科學的偏差管理程序,“無法可依”���;調(diào)查報告的設計沒有按SOP里描述的要求進行呈現(xiàn)���,“執(zhí)法不嚴”�;調(diào)查過程不參照SOP的要求執(zhí)行�����,“有法不依”。
偏差是質(zhì)量保證要素中的一個重要工具�,以上這些常見現(xiàn)象的發(fā)生反映出了背后質(zhì)量管理體系的缺失或無效�����。
4.2調(diào)查報告書寫中存在的問題及建議
4.2.1?常見書寫問題
在偏差報告審核過程中發(fā)現(xiàn)的常見問題主要有:
(1)偏差報告整理不及時�����,不能真實完整地反映出偏差當時情況;
(2)偏差記錄書寫不完整:調(diào)查描述語義混亂�����,沒有主旨�,描述的內(nèi)容和偏差本身關系不大或意思前后存在矛盾�;采取的措施記錄不詳細�、描述不專業(yè)�����;
(3)偏差調(diào)查沒有結論或沒有充足的調(diào)查事實來支持:缺少數(shù)據(jù)和證據(jù)�,結論缺乏說服力���;
(4)結論模糊�,有歧義:使查看報告的人員不能得出調(diào)查人的結論,而是得出其他結論�,即偏差調(diào)查的結論讓人不信服���。
4.2.2?書寫建議
(1)有效性
偏差報告作為偏差實施過程中的原始記錄�����,目的是讓未來的“閱讀者”通過報告能清晰了解整個偏差處理的過程,所以書寫的有效性非常關鍵�。在進行偏差報告書寫前�,應該先明確報告的讀者是參與偏差調(diào)查的團隊(偏差當事人�����、調(diào)查參與者�、上級領導���、批準人)以及未來接受檢查時的審計官或政府法規(guī)部門���。在明白讀者查看報告的目的后,書寫就能切中要點�。
(2)內(nèi)容特性
偏差調(diào)查報告不同于文學寫作�,有其專業(yè)術語和內(nèi)容特性。偏差報告的書寫必須要清晰明白���、簡明扼要地體現(xiàn)出真實性���、相關性�、完整性���、條理性���、一致性�����。
真實性:要根據(jù)可量化和觀測得到的數(shù)據(jù),基于事實進行客觀描述�����,不能受主觀影響���。
相關性:書寫時要使問題焦點保持在偏差涉及的范圍內(nèi),不寫無關的信息或討論�。
完整性:對于必要信息要描述詳細,如用于支持結論的具體事實和數(shù)據(jù)�����、工藝和產(chǎn)品的背景信息等必須描述完整�����。
條理性:在描述開頭就說明主要問題;控制住一個句子中要表達意思的數(shù)量�����,可以使用各種標點符號區(qū)分不同的意思等���。
一致性:在整個報告中同一名稱的專業(yè)稱呼要保持一致,并與法規(guī)或SOP中的用詞一致�����,不要使用方言或俗語�;結論與分析結果、因果關系�����、支持理由�、邏輯等要保持一致���;數(shù)據(jù)和圖表最好采用一致的格式。
5�����、總結
偏差管理是個持續(xù)的過程。只要有人參與的行為就不可避免會有人犯錯�����,所以偏差是極難被消滅掉的�,但是通過將偏差管理落到實處���,能有效減少偏差的發(fā)生�����。質(zhì)量人員既不能對偏差談虎色變���、矯枉過正,也不能掉以輕心�����,漠視已經(jīng)發(fā)生的偏離���。建立適合研發(fā)的偏差管理程序,并在日常實踐中嚴格執(zhí)行�����,定能加速研發(fā)質(zhì)量體系的建立和完善�。
參考文獻
[1]國家食品藥品監(jiān)督管理總局.關于開展藥物臨床試驗數(shù)據(jù)自查核查工作的公告[EB/OL].(2015-07-22).https://www.nmpa.gov.cn/yaopin/ypggtg/ypqtgg/20150722173601172.html.
[2]國家藥品監(jiān)督管理局.仿制藥質(zhì)量和療效一致性評價研制現(xiàn)場核查指導原則[EB/OL].(2017-05-18).https://www.nmpa.gov.cn/yaopin/ypggtg/ypqtgg/20170518144401598.html.
[3]中共中央辦公廳�,國務院辦公廳.關于深化審評審批制度改革鼓勵藥品醫(yī)療器械創(chuàng)新的意見[EB/OL].(2017-10-08).http://www.gov.cn/zhengce/2017-10/08/content_5230105.htm.
[4]全國人民代表大會常務委員會.中華人民共和國藥品管理法[EB/OL].(2019-08-27).https://www.nmpa.gov.cn/xxgk/fgwj/flxzhfg/20190827083801685.html
[5]汪達���,張寶月�����,龍華燕.藥品生產(chǎn)偏差管理討論[J].化工與醫(yī)藥工程,2019���, 40(04):61-64.
[6]厲潔���,黃浩.藥品生產(chǎn)的偏差調(diào)查與分析[J].內(nèi)蒙古中醫(yī)藥,2012���,31(06):66-68.
[7]國際人用藥品注冊技術協(xié)調(diào)會.藥物質(zhì)量體系 Q10[EB/OL].(2008-06-04)https://www.cde.org.cn/ichWeb/guideIch/toGuideIch/1/0.
[8]國際人用藥品注冊技術協(xié)調(diào)會.活性藥物成分(API)的GMP指南 Q7[EB/OL].(2000-11)https://www.cde.org.cn/ichWeb/guideIch/toGuideIch/1/0.
[9]國家食品藥品監(jiān)督管理局藥品認證管理中心.藥品GMP指南 質(zhì)量管理體系[M].北京�����,中國醫(yī)藥科技出版社�,2011:2.
[10]中華人民共和國衛(wèi)生部.藥品生產(chǎn)質(zhì)量管理規(guī)范(2010年修訂)[EB/OL].(2011-01-17).https://www.gov.cn/gongbao/content/2011/content_1907093.htm.
[11]喬曉芳.淺析藥品生產(chǎn)偏差管理進展及改進措施[J].流程工業(yè)���,2018(04):24-27.
[12]國家藥品監(jiān)督管理局.國家藥監(jiān)局關于發(fā)布《藥品生產(chǎn)質(zhì)量管理規(guī)范(2010年修訂)》臨床試驗用藥品附錄的公告(2022年第43號)[EB/OL].(2022-05-27).https://www.nmpa.gov.cn/xxgk/fgwj/xzhgfxwj/20220527182006196.html.
[13]尚海賓.淺析藥品生產(chǎn)設備清潔風險管理[J].流程工業(yè)�,2016(07):49-50.
[14]喬曉芳,楊勝亞�,王志超.藥品生產(chǎn)質(zhì)量風險管理現(xiàn)狀分析及改進措施[J].化工與醫(yī)藥工程,2019���,40(02):53-58.
[15]譚宏宇,單化峰�����,等.FTA法與FMECA法在藥品質(zhì)量偏差調(diào)查中的應用[J].中國藥房�,2016���,27(31):4325-4328.
[16]尚海賓.小容量注射劑干燥及包裝生產(chǎn)質(zhì)量風險管理[J].流程工業(yè),2016(19):48-50.
[17]劉知音�,趙紅菊.藥品生產(chǎn)偏差管理現(xiàn)狀調(diào)研及分析[J].中國藥房�,2011�,22(01):1-4.
本文作者邵玲、葉剛���,第一作者邵玲就職于山東宏濟堂制藥集團股份有限公司、第二作者葉剛就職于鄭州萊士血液制品有限公司�����,僅供交流學習�����。