摘 要 Abstract
為鼓勵創(chuàng)新,加快新藥研發(fā)����,國家藥品監(jiān)督管理局藥品審評中心組織起草制定了《化學(xué)藥品創(chuàng)新藥Ⅲ期臨床試驗前會議藥學(xué)共性問題及相關(guān)技術(shù)要求(試行)》,并正式發(fā)布。本文結(jié)合國內(nèi)創(chuàng)新藥Ⅲ期臨床試驗前藥學(xué)方面溝通會議審評中發(fā)現(xiàn)的問題�,對Ⅲ期臨床試驗前藥學(xué)共性問題進(jìn)行分析總結(jié)����,幫助申請人更好地理解并運用該技術(shù)要求,提高創(chuàng)新藥藥學(xué)溝通交流的質(zhì)量和效率���。
To encourage innovation and accelerate new drug development, the Center for Drug Evaluation has initiated the drafting and official release of the Pharmaceutical Common Issues and Related Technical Requirements in Pre-phase III Clinical Trial Meetings for Innovative Chemical Drugs (Trial). Drawing from issues identified in the review of pre-phase Ⅲ clinical trial meetings of pharmaceutical communication for innovative drugs in China, this paper analyzes and summarizes the recurring pharmaceutical common issues in such trials. The aim is to enhance applicants’ understanding and utilization of these technical requirements, thereby improving the quality and efficiency of pharmaceutical communication concerning innovative drugs.
關(guān)鍵詞 Key words
創(chuàng)新藥���;Ⅲ期臨床試驗�;藥學(xué)共性問題;技術(shù)要求
innovative drugs; phase III clinical trial; pharmaceutical common issues; technical requirements
2020 年7 月1 日實施的《藥品注冊管理辦法》明確指出���,申請人在藥物臨床試驗的關(guān)鍵階段,可以就重大問題與國家藥品監(jiān)督管理局藥品審評中心(以下簡稱藥審中心)進(jìn)行溝通交流[1]���。為加強(qiáng)對藥物研發(fā)與技術(shù)審評溝通交流工作的管理����,藥審中心于2020 年12 月發(fā)布了《藥物研發(fā)與技術(shù)審評溝通交流管理辦法》[2],進(jìn)一步明確了溝通交流的分類及相關(guān)要求����。
為鼓勵新藥研發(fā)和注冊申報,加快新藥上市進(jìn)程�,藥審中心先后發(fā)布了《化學(xué)藥品創(chuàng)新藥Ⅰ期臨床試驗申請藥學(xué)共性問題相關(guān)技術(shù)要求》(2020 年11 月)[3]、《創(chuàng)新藥(化學(xué)藥)臨床試驗期間藥學(xué)變更技術(shù)指導(dǎo)原則(試行)》(2021 年3 月)[4]����、《化學(xué)藥品創(chuàng)新藥上市申請前會議藥學(xué)共性問題及相關(guān)技術(shù)要求》(2021 年11月)[5] 等一系列創(chuàng)新藥藥學(xué)研究相關(guān)指南���。
隨著上述技術(shù)要求和指導(dǎo)原則的發(fā)布�,業(yè)界對Ⅲ期臨床試驗前溝通交流會議相關(guān)技術(shù)要求的呼聲越來越高�。為鼓勵創(chuàng)新,加快創(chuàng)新藥的研發(fā)進(jìn)程�,藥審中心結(jié)合國內(nèi)外法律法規(guī)�、指導(dǎo)原則等,組織起草了《化學(xué)藥品創(chuàng)新藥Ⅲ期臨床試驗前會議藥學(xué)共性問題及相關(guān)技術(shù)要求( 試行)》(以下簡稱《技術(shù)要求》)[6]���,并于2023 年3 月22 日正式發(fā)布���。本文結(jié)合國內(nèi)創(chuàng)新藥Ⅲ期臨床試驗前藥學(xué)溝通會議( 以下簡稱Pre- Ⅲ期藥學(xué)會議)審評中發(fā)現(xiàn)的問題,對Ⅲ期臨床試驗前藥學(xué)共性問題進(jìn)行分析總結(jié),幫助申請人更好地理解并運用《技術(shù)要求》�,提高創(chuàng)新藥藥學(xué)溝通交流的質(zhì)量和效率。
1���、 創(chuàng)新藥溝通交流會議召開情況及發(fā)現(xiàn)的問題
自2015 年藥品審評審批制度改革以來����,國家藥品監(jiān)管部門批準(zhǔn)了多個化學(xué)藥品創(chuàng)新藥上市�,提高了用藥可及性,滿足了臨床用藥需求�。創(chuàng)新藥的臨床期間溝通交流在藥物研發(fā)過程中發(fā)揮了重要作用,申請人與審評團(tuán)隊就重大技術(shù)問題及時溝通����,有助于加快新藥研發(fā)上市。
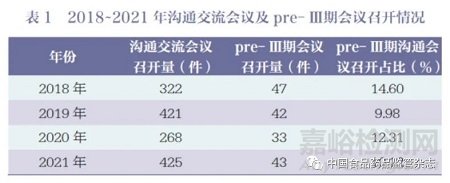
《2021 年度藥品審評報告》指出�,2021 年藥審中心接收溝通交流會議申請4450 件,同比增長37.81% �;辦理溝通交流會議申請3946 件, 同比增長61.00%�。在藥物研發(fā)關(guān)鍵階段召開的Ⅱ類會議占比69.23%,其中���,新藥臨床前(Pre-IND)申請辦理占比32.84%���,新藥生產(chǎn)前(Pre-NDA) 申請辦理占比11.05%�,完成Ⅱ期臨床后(即pre- Ⅲ期溝通交流)申請辦理量為308 件���, 辦理占比7.81%[7]���。2018~2021 年溝通交流會議及pre- Ⅲ期會議召開情況見表1。
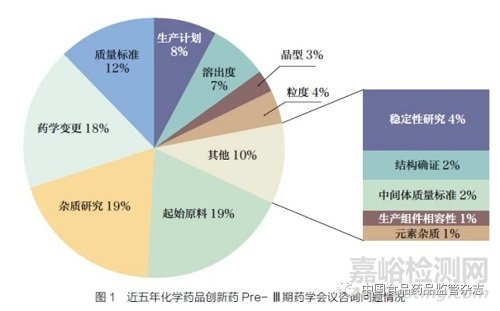
在《技術(shù)要求》起草初期����,起草小組調(diào)研了近五年審評部門承辦的42 個國內(nèi)化學(xué)藥品創(chuàng)新藥Pre- Ⅲ期藥學(xué)會議召開情況,包括召開面對面溝通交流���、視頻會議���、電話會議及書面答復(fù)。這些案例中����,包含179 個咨詢問題����,除去政策法規(guī)方面的24 個問題,其他155 個問題中����,討論頻率較高的問題包括起始原料選擇、雜質(zhì)研究�、藥學(xué)變更等。其他問題包括穩(wěn)定性研究�、結(jié)構(gòu)確證、中間體質(zhì)量標(biāo)準(zhǔn)�、生產(chǎn)組件相容性等,占總問題的10%�,見圖1。調(diào)研過程中���,起草小組發(fā)現(xiàn)�,隨著《創(chuàng)新藥(化學(xué)藥)臨床試驗期間藥學(xué)變更技術(shù)指導(dǎo)原則(試行)》的發(fā)布實施����,2021~2022年的Pre- Ⅲ期藥學(xué)會議中關(guān)于藥學(xué)變更的溝通問題呈下降趨勢���。
調(diào)研發(fā)現(xiàn),Pre- Ⅲ期藥學(xué)會議中存在如下問題:①申請人對Pre- Ⅲ期藥學(xué)會議重視程度不夠����,未提出藥學(xué)溝通申請。②Ⅲ期臨床試驗前或Ⅲ期臨床試驗階段亟待解決的問題�,如起始原料的選擇�,直至完成Ⅲ期臨床試驗后仍懸而未決���,影響藥品上市申請進(jìn)度。③藥學(xué)研究深度不夠���,開發(fā)計劃前瞻性不夠���,Ⅲ期臨床試驗期間仍然有重大藥學(xué)變更,影響Ⅲ期臨床試驗結(jié)果的科學(xué)性�。④提出的藥學(xué)溝通內(nèi)容不具體,提交的支持性研究資料不充分�,影響溝通交流質(zhì)量。
我國醫(yī)藥產(chǎn)業(yè)升級進(jìn)程不斷加快���,新技術(shù)����、新方法�、新標(biāo)準(zhǔn)加速迭代�,創(chuàng)新藥申報數(shù)量的急劇增加,藥物從研發(fā)到上市的進(jìn)程加快�,良好的溝通交流愈發(fā)顯得重要。溝通交流作為審評團(tuán)隊與申請人的重要溝通渠道���,有助于加快藥物研發(fā)上市����。
2���、 Pre-Ⅲ期藥學(xué)溝通交流的總體考慮
創(chuàng)新藥Ⅲ期臨床試驗是用于支持上市的核心安全有效性證據(jù)的確證性臨床試驗�,是藥物的關(guān)鍵臨床試驗���。Ⅲ期臨床試驗階段也是積累工藝認(rèn)知����,加深對產(chǎn)品理解���,制定商業(yè)化生產(chǎn)控制策略的重要階段����。創(chuàng)新藥Pre- Ⅲ期藥學(xué)會議為創(chuàng)新藥研究期間的重要藥學(xué)溝通交流會議,主要目的是解決關(guān)鍵臨床試驗開展前藥學(xué)方面的重大技術(shù)問題����。
《技術(shù)要求》作為創(chuàng)新藥研發(fā)及注冊期間溝通交流技術(shù)要求的一部分,主要討論藥學(xué)共性問題及相關(guān)技術(shù)要求�, 為創(chuàng)新藥Pre- Ⅲ期藥學(xué)會議提供指導(dǎo)?���!都夹g(shù)要求》適用于化學(xué)創(chuàng)新藥和改良型新藥。
申請人為創(chuàng)新藥研發(fā)和注冊申報的責(zé)任主體�,對藥物研發(fā)和注冊申報負(fù)主體責(zé)任。一方面���,基于藥物自身特點及創(chuàng)新藥階段性和漸進(jìn)性特點���,結(jié)合藥物臨床試驗進(jìn)展情況,合理制定藥學(xué)開發(fā)策略和開發(fā)計劃����。另一方面,申請人作為主體責(zé)任人,對品種的認(rèn)識是最深入的�,在開展溝通交流前,全面梳理藥學(xué)研究結(jié)果���,有助于查漏補缺�。開展Ⅲ期臨床試驗前���,藥學(xué)研究需滿足《創(chuàng)新藥(化學(xué)藥)Ⅲ期臨床試驗藥學(xué)研究信息指南》的相關(guān)要求?���!都夹g(shù)要求》在向社會征求意見時,業(yè)界對準(zhǔn)備詳細(xì)的研究資料和研究數(shù)據(jù)提出了較多疑問���,需要說明的是�,為保證溝通交流的質(zhì)量及效率�,建議申請人準(zhǔn)備詳細(xì)的研究資料和研究數(shù)據(jù),資料越詳實���,越有助于解決申請人提出的問題���。另外,建議提出的問題應(yīng)盡量具體、有針對性���。
在Pre- Ⅲ 期藥學(xué)會議中����,涉及工藝復(fù)雜的原料藥或復(fù)雜制劑的溝通問題較多���,情況也比較復(fù)雜�,除《技術(shù)要求》中提及的問題外�,還包括工藝控制、臨床期間的質(zhì)量橋接研究等����。為保證Ⅲ期臨床試驗用樣品的質(zhì)量,支持Ⅲ期臨床試驗的開展及上市申請�,需要開展更多的研究工作?��;谏鲜隹紤]�,建議這一類復(fù)雜品種在Ⅲ期臨床試驗前一定要與監(jiān)管機(jī)構(gòu)進(jìn)行溝通���。
通常�,附條件批準(zhǔn)上市藥品的藥學(xué)、藥理毒理學(xué)要求與常規(guī)批準(zhǔn)上市藥品相同[8]���。對于臨床研究進(jìn)展較快���,預(yù)期臨床有明顯獲益,擬采用藥品加快上市注冊程序(如附條件批準(zhǔn)程序等)的創(chuàng)新藥����,或者臨床開發(fā)周期比較短的創(chuàng)新藥�,申請人需盡早開展藥學(xué)研究,藥學(xué)研究計劃需與臨床研究計劃相匹配�,藥學(xué)研究進(jìn)度不應(yīng)成為影響藥物上市的限速步驟。
3���、 常見的藥學(xué)共性問題及審評考慮
3.1 起始原料選擇
起始原料選擇是Pre- Ⅲ期藥學(xué)會議中非常重要的討論問題���。審評中發(fā)現(xiàn),有些申請人并未重視這個問題���,導(dǎo)致在上市申請前溝通交流或上市申請時要求前延合成路線����,影響注冊核查工作的開展,進(jìn)而影響藥品上市進(jìn)程����。
如果產(chǎn)品計劃采用藥品加快上市注冊程序(如附條件批準(zhǔn)程序等),需在用于支持上市的關(guān)鍵Ⅱ期臨床試驗開展前與審評機(jī)構(gòu)討論起始原料的選擇問題�。
審評工作中發(fā)現(xiàn),起始原料的選擇經(jīng)常會遇到如下問題:①擬定起始原料選擇依據(jù)不充分���。②提供的雜質(zhì)分析不全面���。③起始原料中引入的雜質(zhì)在后續(xù)工藝步驟匯總的清除轉(zhuǎn)化研究不全面,無法支持目前起始原料的選擇�。
起始原料的選擇需按照ICHQ11[9] 及問答文件[10] 提出的基本原則開展研究:①起始原料應(yīng)當(dāng)具備明確的化學(xué)特性和結(jié)構(gòu)����,未分離的中間體通常不被考慮作為合適的起始原料�。②起始原料作為重要的結(jié)構(gòu)片段并入原料藥的結(jié)構(gòu)中�。③通用技術(shù)文件(CTD)“3. 2. S. 2. 2”應(yīng)包含對原料藥的雜質(zhì)譜產(chǎn)生影響的生產(chǎn)步驟�。④應(yīng)充分描述原料藥的生產(chǎn)工藝,明確雜質(zhì)在工藝過程中的產(chǎn)生���、去向與清除�,控制策略滿足雜質(zhì)控制要求。⑤采用匯聚型原料藥生產(chǎn)工藝的每個分支開始于一個或多個起始原料���。每個分支從首次使用起始原料的步驟開始���,需要遵循生產(chǎn)質(zhì)量管理規(guī)范����,并結(jié)合適當(dāng)?shù)目刂撇呗詾樵纤幍馁|(zhì)量提供保證���。⑥在生產(chǎn)工藝開始階段附近發(fā)生的物料屬性或操作條件的改變對原料藥質(zhì)量的潛在影響較小����。需要特別說明的是,在選擇起始原料時應(yīng)當(dāng)考慮上述全部原則,而不是孤立地嚴(yán)格遵循每一個原則。
在論證起始原料選擇合理性時�,可以提供包括但不限于以下資料作為支持性依據(jù):①起始原料的雜質(zhì)譜分析是否全面�,分析方法檢測起始原料中雜質(zhì)的可行性����,是否經(jīng)過適當(dāng)驗證���。②雜質(zhì)及其衍生物在后續(xù)工藝中的去向和清除研究情況����。③起始原料的質(zhì)量標(biāo)準(zhǔn)如何實現(xiàn)整體的質(zhì)量控制策略����,可以結(jié)合中間體雜質(zhì)研究情況論證起始原料的質(zhì)量控制對終產(chǎn)品控制策略的影響�。
ICH Q11 及問答文件闡述了半合成原料藥的起始原料選擇的相關(guān)考慮。通常情況下����,應(yīng)從源物質(zhì)(微生物或植物)開始描述生產(chǎn)工藝?��?紤]到發(fā)酵/ 提取工藝及純化工藝比較復(fù)雜�,產(chǎn)品組分比例和雜質(zhì)水平受工藝影響較大,建議將微生物發(fā)酵/ 提取步驟納入原料藥生產(chǎn)工藝����,并將微生物發(fā)酵/ 提取及分離純化工藝納入原料藥生產(chǎn)質(zhì)量管理體系。ICHQ11 指出�,如果能夠證明合成工藝中的一個分離中間體符合上述化學(xué)合成原料藥起始原料的選擇原則,則該分離中間體可被提議作為起始原料�。此種情況,建議申請人盡早與監(jiān)管機(jī)構(gòu)進(jìn)行討論���。
在審評中發(fā)現(xiàn)����,申請人由于原料供應(yīng)等問題����,會選擇兩家或多家起始原料生產(chǎn)商�,但相關(guān)研究并不充分����,不能證明不同生產(chǎn)商的起始原料具有可替代性����。不同來源的起始原料的研究可參考《已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》[11] 的相關(guān)要求開展���。根據(jù)生產(chǎn)商的制備工藝,并結(jié)合雜質(zhì)的清除轉(zhuǎn)化研究情況,合理制定起始原料的質(zhì)量控制策略�。
3.2 有關(guān)物質(zhì)
創(chuàng)新藥的研發(fā)具有階段性和漸進(jìn)性的特點����,隨著臨床試驗的進(jìn)展�,對藥物特性(原料藥的合成工藝及降解途徑)的認(rèn)知不斷加深���,原料藥及制劑的雜質(zhì)譜分析會逐漸完善�。如何保證有關(guān)物質(zhì)分析方法能有效分離和檢出潛在雜質(zhì)�?通常情況下���,有關(guān)物質(zhì)分析方法的開發(fā)是在雜質(zhì)譜分析全面的基礎(chǔ)上���,結(jié)合化合物及雜質(zhì)的理化性質(zhì)���,采用不同分離原理的分析方法���,如高效液相色譜法���、氣相色譜法���、離子交換色譜法、分子排阻色譜法等���,進(jìn)一步證明方法對潛在雜質(zhì)的分離和檢出能力���;對于采用液相色譜法,需對色譜柱����、檢測波長、洗脫程序等進(jìn)行篩選����;用于方法開發(fā)的樣品需具有一定的代表性�,結(jié)合產(chǎn)品特性����,可以從定向制備的雜質(zhì)���、含一定雜質(zhì)的粗品或粗品母液(如5%~10% 雜質(zhì)含量)����、適度降解的樣品���、加速和長期穩(wěn)定性樣品等樣品中進(jìn)行選擇���。如果制劑工藝中用到熱熔擠出、高溫滅菌條件(如121℃����、15min 滅菌)等工藝,需考慮有關(guān)物質(zhì)方法能否對該苛刻條件下產(chǎn)生的潛在降解雜質(zhì)具有較好的分離和檢出能力���。
有關(guān)物質(zhì)是藥物的關(guān)鍵質(zhì)量屬性�。臨床試驗期間,申請人需不斷積累雜質(zhì)的檢測數(shù)據(jù)���。如果有關(guān)物質(zhì)分析方法發(fā)生變更����,需關(guān)注不同方法間有關(guān)物質(zhì)檢測結(jié)果的橋接研究����。需要說明的是,雖然ICH Q3A[12]����、ICH Q3B[13]不適用于臨床試驗期間, 但鼓勵申請人參考ICH Q3A����、ICHQ3B 等指導(dǎo)原則對超過鑒定限的雜質(zhì)、穩(wěn)定性期間出現(xiàn)的超過鑒定限的雜質(zhì)進(jìn)行歸屬鑒別����,各雜質(zhì)水平不得超過已有的動物安全性試驗支持水平。雜質(zhì)清除轉(zhuǎn)化研究是支持雜質(zhì)控制策略的一項重要研究內(nèi)容����,申請人可以根據(jù)臨床試驗進(jìn)展開展此項研究����。
3.3 致突變雜質(zhì)
前期調(diào)研中����,Pre- Ⅲ期藥學(xué)溝通案例提出致突變雜質(zhì)問題的比例約為7%����。通常情況下����,致突變雜質(zhì)的研究需參照ICH M7[14]開展并制定控制策略。用于晚期腫瘤患者的藥物可參照ICH M7和ICH S9[15] 進(jìn)行致突變雜質(zhì)的評估和控制�。如果早期開發(fā)用于晚期腫瘤,隨著臨床研究的進(jìn)展和開發(fā)計劃���,如擬開發(fā)適應(yīng)癥為非晚期腫瘤(如一線用藥)或其他非腫瘤適應(yīng)癥����,需重新評估擬定的致突變雜質(zhì)控制策略是否還適用�。
另外,藥審中心于2020 年發(fā)布了《化學(xué)藥物中亞硝胺類雜質(zhì)研究技術(shù)指導(dǎo)原則(試行)》[16]�,美國食品藥品監(jiān)督管理局(FDA)于2021 年發(fā)布了亞硝胺雜質(zhì)的控制指導(dǎo)原則[17]����,歐洲藥品管理局(EMA) 分別于2020 年���、2023 年發(fā)布了亞硝胺雜質(zhì)的評估報告[18] 及問答文件[19]����。申請人在開展亞硝胺雜質(zhì)研究過程中����,可以參考上述指導(dǎo)原則進(jìn)行評估,制定合理的控制策略�。
3.4 晶型
對于固體制劑、半固體制劑���、混懸劑等劑型來說����,晶型對制劑生產(chǎn)工藝����、穩(wěn)定性以及制劑的溶出行為、藥代動力學(xué)和療效均可能存在影響����。
在早期產(chǎn)品開發(fā)時需開展晶型研究�,包括晶型篩選����、晶型溶解度與溶解速率、結(jié)晶工藝開發(fā)���、晶型穩(wěn)定性等����。需說明選擇目標(biāo)晶型的依據(jù)����,以及在原料藥粉碎���、制劑生產(chǎn)及貯藏過程中晶型的穩(wěn)定情況���。原料藥經(jīng)干燥、粉碎�、制粒和壓片等制劑工藝步驟,在溫度和濕度等環(huán)境因素的作用下����,均可能出現(xiàn)轉(zhuǎn)晶現(xiàn)象����,進(jìn)而影響制劑的溶出行為和(或)生物利用度���。因此,通常采用熱力學(xué)穩(wěn)定的晶型進(jìn)行開發(fā)�,注意避免采用混晶或生產(chǎn)、貯藏過程中發(fā)生轉(zhuǎn)晶后形成混晶�。
建議參照ICH Q11 等指導(dǎo)原則����,加強(qiáng)原料藥結(jié)晶工藝研究及生產(chǎn)工藝參數(shù)的控制���,保持原料藥批間晶型的一致性。
3.5 原料藥的粒度
原料藥的粒度是原料藥的一項關(guān)鍵質(zhì)量屬性�,特別是難溶性的藥物,原料藥粒度對制劑的混合均勻度���、溶出行為及生物利用度均有重要影響�。一方面,可通過加強(qiáng)原料藥結(jié)晶����、粉碎步驟工藝參數(shù)的控制,包括但不限于結(jié)晶溶劑的用量/ 比例���、結(jié)晶溫度����、升降溫速率����、粉碎參數(shù)等����,從而控制原料藥粒度在擬定范圍內(nèi)���。另一方面����,粒度測定方法應(yīng)專屬����、靈敏�,必要時可考慮與顯微鏡檢測結(jié)果進(jìn)行對比�。臨床試驗期間需持續(xù)積累粒度檢測數(shù)據(jù)�,結(jié)合粒度對制劑關(guān)鍵質(zhì)量屬性(如混合均勻度���、溶出行為等)及體內(nèi)暴露行為的影響�,合理制定控制策略。
3.6 溶出度/ 釋放度研究
溶出度/ 釋放度是口服固體制劑的一項關(guān)鍵質(zhì)量屬性����。擬定的溶出度/ 釋放度方法需有適度的區(qū)分力。溶出度/ 釋放度方法開發(fā)時���,建議根據(jù)藥物在37℃的pH- 溶解度曲線,選擇具有適度區(qū)分力的溶出介質(zhì)/ 釋放介質(zhì),并對裝置(槳法/ 籃法等)、介質(zhì)體積���、轉(zhuǎn)速等進(jìn)行篩選����,如需加入表面活性劑�,需論證表面活性劑加入的必要性和加入量的合理性���,對其種類����、來源�、用量等進(jìn)行篩選,并開展方法的區(qū)分力研究�,可以包括對處方變量、工藝參數(shù)變量����、原料藥粒徑、晶型等的區(qū)分力�。Ⅲ期臨床試驗期間需注意積累數(shù)據(jù)。
3.7 批量
《技術(shù)要求》中���,批量問題并未作為單獨的共性問題進(jìn)行討論����,但實際工作中批量問題經(jīng)常出現(xiàn)在溝通交流問題清單中�。通常情況下,普通口服固體制劑的關(guān)鍵臨床批次樣品的批量建議不低于10 萬片/ 粒,如已確定為罕見病用藥�,這類品種的批量可以及時與審評機(jī)構(gòu)進(jìn)行討論確定合理批量。其他劑型的批量可以參照《化學(xué)仿制藥注冊批生產(chǎn)規(guī)模的一般性要求(試行)》[20] 等指導(dǎo)原則來制定。
3.8 臨床試驗期間藥學(xué)變更
臨床試驗期間發(fā)生的藥學(xué)變更可以參照《創(chuàng)新藥(化學(xué)藥)臨床試驗期間藥學(xué)變更技術(shù)指導(dǎo)原則(試行)》開展研究���,并按照《藥品注冊管理辦法》相關(guān)要求�,選擇合適的申報路徑。
需要特別關(guān)注的是,皮膚外用制劑���、復(fù)雜制劑(如微球/ 微乳、脂質(zhì)體�、混懸型注射液等)�,由于其處方工藝���、制備工藝相對比較復(fù)雜,對生產(chǎn)設(shè)備的要求比較高�,如果發(fā)生生產(chǎn)場地、處方工藝����、批量的變更,僅通過藥學(xué)對比研究無法證明變更前后產(chǎn)品質(zhì)量一致�。因此,建議這類復(fù)雜制劑的生產(chǎn)場地���、處方工藝、批量等的變更盡量在關(guān)鍵臨床試驗前開展。上市申請的生產(chǎn)場地����、處方工藝、批量需與Ⅲ期臨床試驗保持一致。
4���、 結(jié) 語
隨著藥品審評審批制度改革的不斷深入����,藥品研發(fā)注冊的配套法規(guī)����、指導(dǎo)原則及技術(shù)要求等不斷完善�,為藥物研發(fā)注冊提供了技術(shù)指導(dǎo)。藥審中心堅持鼓勵以臨床價值為導(dǎo)向的新藥好藥、罕見病用藥�、重大傳染病用藥及公共衛(wèi)生方面的臨床急需藥品的研發(fā)創(chuàng)新����,加速完善藥品審評審批標(biāo)準(zhǔn)體系建設(shè)����。《技術(shù)要求》與《化學(xué)藥品創(chuàng)新藥Ⅰ期臨床試驗申請藥學(xué)共性問題相關(guān)技術(shù)要求》《化學(xué)藥品創(chuàng)新藥上市申請前會議藥學(xué)共性問題及相關(guān)技術(shù)要求》組成了化學(xué)藥品創(chuàng)新藥臨床期間及上市前的溝通交流技術(shù)要求體系�,服務(wù)藥品創(chuàng)新����。申請人可以通過運用這些技術(shù)要求,就重大技術(shù)問題及時提出溝通交流,加快藥品研發(fā)上市����。
引用本文
徐立華����,周浩輝�,趙一飛�,王亞敏*.化學(xué)藥品創(chuàng)新藥Ⅲ期臨床試驗前會議的藥學(xué)共性問題及審評考慮[J].中國食品藥品監(jiān)管.2023.08(235):32-37.