引言
藥品變更管理貫穿研發(fā)、臨床���、生產(chǎn)�����、流通�����、上市后研究等全生命周期�,在創(chuàng)新藥物的臨床研究期間,藥學變更是不可避免的重要環(huán)節(jié)���。在進行藥學變更時���,臨床試驗的安全性和數(shù)據(jù)的可靠性是首要考慮因素。任何藥學變更都需要經(jīng)過科學嚴謹?shù)脑u估和審查�,以確保對患者的治療效果和安全性沒有負面影響。此外���,在藥學變更過程中�����,需要進行必要的數(shù)據(jù)記錄和文檔整理���,確保變更過程的追蹤和監(jiān)控�����,并通過適當程序(補充申請�����、DSUR(研發(fā)階段安全性更新報告))及時通報監(jiān)管部門�?����?偟膩碚f���,創(chuàng)新藥物的臨床期間�,藥學變更是為了不斷優(yōu)化和改進藥物���,提高臨床療效和安全性�����。但在進行藥學變更時���,必須遵循嚴格的規(guī)定和指導,以確保變更的安全性和數(shù)據(jù)的可靠性���。
一���、創(chuàng)新化藥臨床期間與上市后藥學變更管理對比
通過對《創(chuàng)新藥(化學藥)臨床試驗期間藥學變更技術(shù)指導原則(試行)》、《已上市化學藥品藥學變更研究技術(shù)指導原則(試行)》中有關(guān)內(nèi)容的比較�����,可以看出�����,在這兩個文件中�,既有相似之處,又有不同之處�����。兩者都是采取以安全風險評價為基礎�����,由申請者進行風險評估并確定風險等級,但是其管理方法上卻有所不同�。
1.1 變更分類和要求
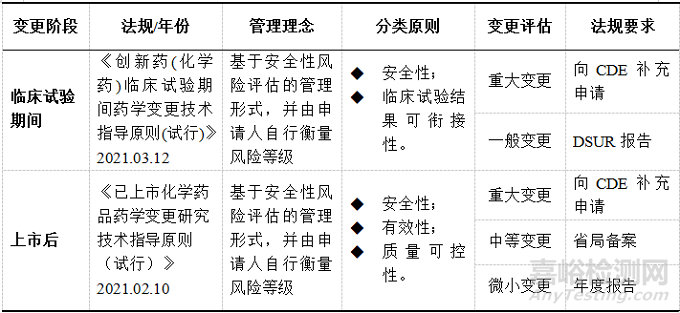
臨床試驗期間藥學變更按照對臨床受試者安全性、臨床試驗結(jié)果可銜接性影響風險分為重大變更���、一般變更兩類進行管理���。上市后藥學變更按照對產(chǎn)品安全性、有效性和質(zhì)量可控性的影響分為重大變更�����、中等變更�、微小變更三類進行管理,見表1�����。
▲表1-創(chuàng)新化藥臨床試驗期間和上市后藥學變更分類和要求
注:早期臨床研究階段�,藥物的人體安全性尚未完全確立,重點評估藥學變更對于受試者安全性可能產(chǎn)生的影響���;關(guān)鍵臨床研究階段�,除需重點關(guān)注受試者安全性外,還需兼顧臨床試驗結(jié)果的可銜接性�����。
1.2 變更程序
申請人評估認為可能增加受試者安全風險的變更���,應當按《藥品注冊管理辦法》提出補充申請,認為不影響受試者安全的�����,可以直接實施并在研發(fā)期間安全性更新報告或在上市后年度報告中說明�。
(1)臨床期間變更
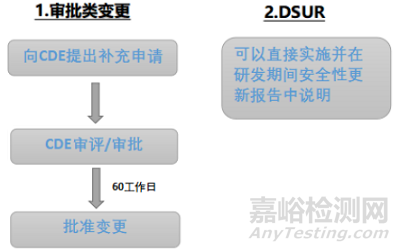
與上市后藥品變更不同,對于臨床試驗期間的藥學變更�����,CDE把評估權(quán)更多的交給了申請人���。只有影響受試者安全性的重大變更要向CDE提交補充申請���,如果臨床期間的藥學變更屬于未影響受試者安全性的一般變更,申請人可以直接實施并在DSUR報告中說明�����,見圖1。
▲圖1-創(chuàng)新化藥臨床試驗期間藥學變更程序
(2)上市后變更
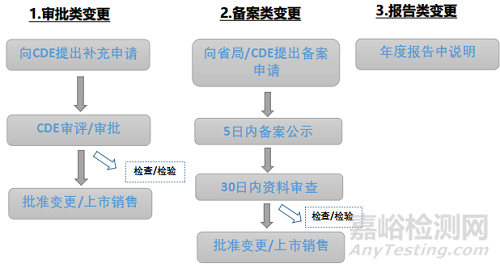
根據(jù)法規(guī)要求藥品上市后變更程序主要包括審批類變更���、備案類變更及報告類變更�����,見圖2�����。
▲圖2-創(chuàng)新化藥上市后藥學變更程序
其中CDE審批類變更申請審評時限為60工作日���,補充申請合并申報事項的,審評時限為80工作日���,其中涉及臨床試驗研究數(shù)據(jù)審查�����、藥品注冊核查檢驗的審評時限為200工作日�����。
1.3 補充申請資料
根據(jù)2021年2月10日國家藥監(jiān)局發(fā)布《已上市化學藥品變更事項及申報資料要求》附件�,國家藥品監(jiān)管部門審批的補充申請事項包括:
國家藥品監(jiān)管部門發(fā)布的已上市化學藥品藥學變更相關(guān)技術(shù)指導原則中屬于重大變更的事項。
國家藥品監(jiān)管部門發(fā)布的已上市化學藥品臨床變更相關(guān)技術(shù)指導原則中屬于重大變更的事項�����。
藥品上市許可持有人主體變更���。
使用藥品商品名。
國家藥品監(jiān)管部門規(guī)定需要審批的其他事項���。
從上述規(guī)定來看創(chuàng)新藥臨床期間和上市后藥學重大變更均納入國家藥品監(jiān)管部門審批補充申請管理事項�。
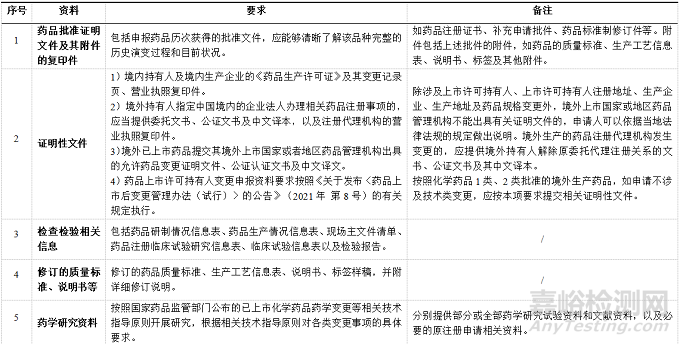
藥品上市許可持有人應根據(jù)所申請事項�,按表2所示,整理并提交申報資料�����,不適用的項目應注明不適用并說明理由�。
▲表2-創(chuàng)新化藥補充申請資料
二、臨床試驗期間藥學變更分類與研究內(nèi)容
創(chuàng)新化藥臨床期間藥學變更包括原料藥和制劑兩大類�。對于原料藥變更,需要考慮生產(chǎn)場地�����、工藝、質(zhì)量標準�����、包裝容器和貯藏條件等�����。而在制劑變更方面���,則需要注意生產(chǎn)場地���、處方工藝、輔料工藝���、劑型與規(guī)格�、質(zhì)量標準�����、包裝容器和貯藏條件等�����。申請人通過詳細的研究和測試,確保變更后的藥品仍滿足臨床試驗的要求���,并保證受試者的安全�����。
2.1 原料藥變更
對于原料藥變更研究的一般原則���,是需結(jié)合其對相應制劑的質(zhì)量影響開展評估和研究�����。針對自身產(chǎn)品特點���,根據(jù)風險評估結(jié)果開展研究�����。如出現(xiàn)變更前后產(chǎn)品質(zhì)量不可比的情況���,申請人需要通過前期的數(shù)據(jù)積累進行風險評估�,合理的完成橋接研究���。
▲表3- 臨床試驗期間原料藥藥學變更分類與研究
對于復雜分子(如:合成多肽���、小分子核酸等)、復雜工藝(如:發(fā)酵類���、生物來源提取物類等)原料藥的變更�����,其對臨床樣品的潛在影響的體外評估手段相比化藥更具特異性���,需在遵照本指導原則研究思路的基礎上,結(jié)合產(chǎn)品自身特點開展充分的風險評估及變更研究工作���,以獲得更多的變更支持性數(shù)據(jù)�。
2.2 制劑變更
對于制劑變更�,需重點從變更對于藥物的制劑性能、安全性相關(guān)指標的影響來展開評估和研究�。
▲表4- 臨床試驗期間制劑藥學變更分類與研究
一般而言���,變更制劑劑型、規(guī)格�����、處方���、生產(chǎn)工藝�����、包裝和貯藏條件等都有可能會對制劑性能產(chǎn)生影響,在變更研究中需結(jié)合藥物性質(zhì)�����、劑型特性�����、處方工藝特點以及變更的具體情況評估變更對制劑性能可能產(chǎn)生的影響���,根據(jù)風險評估結(jié)果���,選擇適宜的制劑性能相關(guān)指標開展變更支持性研究���,必要時還需考慮進行制劑體內(nèi)橋接研究。
對于安全性相關(guān)指標,雜質(zhì)種類和雜質(zhì)水平是影響臨床試驗藥品安全性的重要因素�����。對于注射劑���、吸入制劑�����、植入劑等而言�,還需關(guān)注無菌�、細菌內(nèi)毒素(或熱原)�����、不溶性微粒(真溶液型注射劑)�、可見異物(真溶液型注射劑)、滲透壓摩爾濃度(注射劑)等���。需確保變更后產(chǎn)品的相關(guān)指標仍符合安全性要求���。
三�����、結(jié)束語
申請人在創(chuàng)新化藥臨床期間進行藥學變更研究時�����,申請人需要明確變更的原因、事項和程度�。同時,要結(jié)合品種特點和具體的變更內(nèi)容�,按照風險評估的思路來評估變更可能對藥品質(zhì)量、臨床試驗受試者安全性和臨床試驗結(jié)果可銜接性產(chǎn)生的影響�。在評估的基礎上�����,可以判斷變更是重大變更還是一般變更�����,并相應地展開研究工作。此外���,藥學變更往往不是孤立發(fā)生的�。例如�,在進行生產(chǎn)場地變更時�����,可能伴隨著生產(chǎn)設備和生產(chǎn)工藝的變更;在進行處方變更時�,可能引發(fā)藥品質(zhì)量標準的變更或與之相關(guān)�����。對于多個變更同時發(fā)生且存在關(guān)聯(lián)的情況,可以根據(jù)《創(chuàng)新藥(化學藥)臨床試驗期間藥學變更技術(shù)指導原則(試行)》的基本思路�,分別展開研究工作。重點關(guān)注技術(shù)要求較高的變更類別�,并注意多個變更可能帶來的疊加影響。
總之�,進行藥學變更研究時���,申請人需全面評估變更的影響�,確定變更的風險等級�����,合法�、合規(guī)地展開相應的研究工作���,確保變更后生產(chǎn)的藥品符合質(zhì)量要求�、保證臨床試驗受試者的安全���,并能夠產(chǎn)生可靠的臨床試驗結(jié)果�。
參考文獻
[1] 《創(chuàng)新藥(化學藥)臨床試驗期間藥學變更技術(shù)指導原則(試行)》(2021年第22號).CDE,2021-03-12.
[2] 《已上市化學藥品藥學變更研究技術(shù)指導原則(試行)》 (2021年第16號).CDE�,2021-02-10.
[3] 《藥品注冊管理辦法》(2020年第27號). 國家市場監(jiān)督管理總局�����,2020-04-07.
[4] 《已上市化學藥品變更事項及申報資料要求》 (2021年第15號).國家藥品監(jiān)督管理局�����,2021-02-10.