通過探討《組合產(chǎn)品上市前路徑指導(dǎo)原則》的相關(guān)要求,分析美國食品藥品管理局(Food and DrugAdministration, FDA)對含藥醫(yī)療器械5種典型情況的申報(bào)路徑���、參比器械選擇的審評關(guān)注點(diǎn)�����,以期對我國以醫(yī)療器械作用為主的藥械組合產(chǎn)品的申報(bào)注冊提供參考借鑒���。
本研究通過探討美國食品藥品管理局(Foodand Drug Administration, FDA)《組合產(chǎn)品上市前路徑指導(dǎo)原則》(Principles of Premarket Pathwaysfor Combination Products Guidance for Industry andFDA Staff)[1]中對以醫(yī)療器械作用為主的組合產(chǎn)品(device-led combination product,以下簡稱“含藥醫(yī)療器械”)上市前申報(bào)路徑及參比器械選擇的審評關(guān)注點(diǎn)���,結(jié)合我國該類產(chǎn)品的審評情況��,分析其借鑒意義�����,供我國藥械組合產(chǎn)品相關(guān)監(jiān)管部門和從業(yè)人員參考和借鑒���。
1�����、 FDA《組合產(chǎn)品上市前路徑指導(dǎo)原則》 內(nèi)容概要
2019年2月��,美國FDA發(fā)布了《組合產(chǎn)品上市前路徑指導(dǎo)原則》草案稿���,歷時(shí)近3年,于2022年1月發(fā)布了該指導(dǎo)原則的正式稿�����。該指導(dǎo)原則闡明了FDA對組合產(chǎn)品上市前申報(bào)路徑選擇原則的考量���,包括申請人與FDA各審評機(jī)構(gòu)��、組合產(chǎn)品辦公室之間的溝通和協(xié)調(diào)���,組合產(chǎn)品的上市前監(jiān)管基礎(chǔ),以醫(yī)療器械作用為主的組合產(chǎn)品(deviceledcombination product)��、以藥品作用為主的組合產(chǎn)品(drug-led combination product)��、以生物制品作用為主的組合產(chǎn)品(biologic-led combinationproduct)各自的上市前申請路徑類型以及選擇恰當(dāng)?shù)纳暾埪窂剿杩紤]的關(guān)鍵因素等內(nèi)容��。
該指導(dǎo)原則在第Ⅲ部分“組合產(chǎn)品的上市前監(jiān)管基礎(chǔ)”中明確指出���,F(xiàn)DA認(rèn)為藥品��、器械和生物制品在作為組合產(chǎn)品的組成部分時(shí)���,保留各自獨(dú)立的監(jiān)管身份,即在其作為組合產(chǎn)品的組成部分時(shí)���,仍適用于藥品��、醫(yī)療器械和生物制品的法規(guī)和監(jiān)管要求���。因此,對組合產(chǎn)品上市前的整體安全性和有效性要求�����,源自組合產(chǎn)品各組成部分相對應(yīng)的、適用的法規(guī)和監(jiān)管要求���。FDA另行制定組合產(chǎn)品的專項(xiàng)監(jiān)管要求���,通常是為了解決組合產(chǎn)品的藥品���、器械和生物制品不同組成部分的法規(guī)和監(jiān)管要求之間的重疊問題和不同監(jiān)管領(lǐng)域的差別問題��。
同時(shí)�����,F(xiàn)DA在該指導(dǎo)原則的附錄部分指出���,由于申請人對組合產(chǎn)品申請途徑選擇存在疑問的情況最集中出現(xiàn)在含藥醫(yī)療器械,因此在附錄部分以含藥醫(yī)療器械的5種典型情況為實(shí)例進(jìn)行說明�����,介紹了在產(chǎn)品注冊申報(bào)時(shí),含藥醫(yī)療器械如何選擇合適的上市前申報(bào)路徑及如何選擇恰當(dāng)?shù)膮⒈绕餍档葍?nèi)容�����。
2��、 含藥醫(yī)療器械5種典型情況如何選擇產(chǎn)品的申報(bào)路徑及參比器械
在FDA,含藥醫(yī)療器械按其風(fēng)險(xiǎn)程度�����、新型程度等不同情況,上市前申請路徑主要分為上市前批準(zhǔn)(premarket approval application, PMA)�����、新型醫(yī)療器械申請(De Novo)以及上市前通知[510(k)submission]這3種類型?��!督M合產(chǎn)品上市前路徑指導(dǎo)原則》附錄部分以含抗菌劑的腹腔引流管為例,給出了抗菌涂層首次添加到已明確管理類別的醫(yī)療器械�����、組合產(chǎn)品的藥品部分增加新適應(yīng)證、組合產(chǎn)品上的藥物涂層方法發(fā)生改變���、組合產(chǎn)品藥品部分的濃度降低�����、組合產(chǎn)品的藥品部分發(fā)生改變(用其他抗菌劑替代其組合產(chǎn)品原有的藥品部分)等5種典型情況的具體說明。
2.1 典型情況1
典型情況1為抗菌涂層首次添加到已明確管理類別的醫(yī)療器械���。假設(shè)參比產(chǎn)品(predicateproduct)為不含藥品或生物制品���、已被界定為Ⅱ類醫(yī)療器械���、歸屬510(k)管理的產(chǎn)品(如腹腔引流管);假設(shè)組合產(chǎn)品所含的抗菌涂層(抗菌劑A)��,是與已獲批用于靜脈給藥的新藥申請(newdrug application, NDA)藥物具有相同的活性成分���,該成分用于治療急性細(xì)菌性皮膚病和皮膚感染的療效和風(fēng)險(xiǎn)已被確認(rèn);申請人已向FDA提供引用NDA授權(quán)的文件[根據(jù)美國聯(lián)邦法案第503(g)(5)節(jié)���,在滿足503(g)(5)(A)&(C)所有要求的前提下���,可沿用FDA先前對抗菌劑A的NDA安全性和有效性的審查結(jié)果]���。
申請人擬將抗菌涂層(抗菌劑A)添加到上述參比產(chǎn)品中��,制成單一實(shí)體的組合產(chǎn)品(以下簡稱“產(chǎn)品A”)���,在器械上添加抗菌涂層的目的是防止與外科手術(shù)及器械使用相關(guān)的感染���,并且該抗菌成分以往未與該類器械進(jìn)行組合�����。
對于這種典型情況��,F(xiàn)DA認(rèn)為雖然參比產(chǎn)品和組合產(chǎn)品的使用目的相同��,但組合產(chǎn)品上添加抗菌涂層��,產(chǎn)生了新的預(yù)期用途�����,且二者技術(shù)特征存在差異��,引起了影響產(chǎn)品安全有效性的新問題��,因此二者不具有實(shí)質(zhì)等同性(not substantiallyequivalent, NSE)���。在這種情況下��,F(xiàn)DA不允許組合產(chǎn)品中藥品部分單獨(dú)申請采用510(k)實(shí)質(zhì)等同途徑的方式。組合產(chǎn)品與參比產(chǎn)品的比較不能充分證明組合產(chǎn)品的藥品部分在其新使用條件下的安全有效性(包括新的藥品適應(yīng)證�����、給藥途徑以及藥品與器械的組合使用)���。
若組合產(chǎn)品符合美國聯(lián)邦法案第513(a)(1)(A)或(B)和513(f)(2)節(jié)的要求,則該組合產(chǎn)品可能適合申請De Novo途徑��。在確定是否獲準(zhǔn)De Novo申請時(shí)�����,若申請人已被授權(quán)可引用藥品NDA申請中的數(shù)據(jù)���,F(xiàn)DA在審查該組合產(chǎn)品的De Novo申請時(shí),會(huì)考慮將相關(guān)的藥品NDA申請數(shù)據(jù)作為支持資料。若該組合產(chǎn)品不符合De Novo途徑申報(bào)的要求��,則需要按PMA途徑申報(bào)。
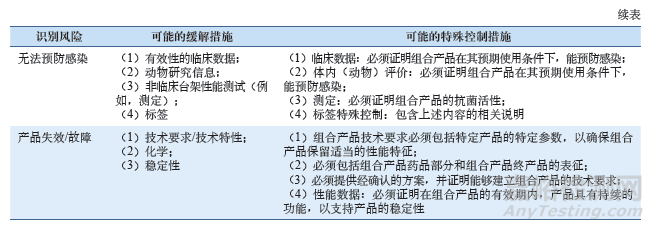
若該組合產(chǎn)品符合De Novo途徑申報(bào)的要求���,F(xiàn)DA在其風(fēng)險(xiǎn)識(shí)別和緩解�����、控制措施部分主要關(guān)注的問題�����,如表1所示���。
2.2 典型情況2
典型情況2為組合產(chǎn)品的藥品部分增加新適應(yīng)證。假設(shè)參比產(chǎn)品為已上市的產(chǎn)品A�����;新組合產(chǎn)品的藥品部分與產(chǎn)品A的藥品部分相同��;申請人已向FDA提供引用NDA授權(quán)的文件��。基于藥品部分的藥理學(xué)性質(zhì)��,除抗菌特性用途之外���,申請人提出產(chǎn)品A還具有新的抗感染適應(yīng)證�����,通過減輕植入后宿主環(huán)境中的感染來提高產(chǎn)品的整體性能��。
對于這種典型情況���,F(xiàn)DA認(rèn)為雖然二者的使用目的相同,新組合產(chǎn)品增加抗感染適應(yīng)證及減輕感染的用途��,引起了安全有效性的新問題���,不利于與參比產(chǎn)品進(jìn)行對比��,二者不具有實(shí)質(zhì)等同性。在這種情況下,不允許510(k)途徑實(shí)質(zhì)等同的方式應(yīng)用于組合產(chǎn)品中藥品部分的單獨(dú)申請。新產(chǎn)品與參比產(chǎn)品(產(chǎn)品A)的比較不能充分證明組合產(chǎn)品藥品部分新適應(yīng)證的安全有效性。
2.3 典型情況3
典型情況3為組合產(chǎn)品上的藥物涂層方法發(fā)生改變��。假設(shè)參比產(chǎn)品為已上市的產(chǎn)品A��;新組合產(chǎn)品與產(chǎn)品A的藥品部分相同��;申請人已向FDA提供引用NDA授權(quán)的文件���。申請人擬改變藥物涂層方式以改進(jìn)產(chǎn)品A(減少藥物從器械上的釋放),目的是防止?jié)撛诘牟涣际录投拘裕瑫r(shí)保持藥物的有效性�����,組合產(chǎn)品的預(yù)期用途或標(biāo)簽不變��。
對于這種典型情況���,F(xiàn)DA認(rèn)為該組合產(chǎn)品的材料、其他特征與參比產(chǎn)品相比發(fā)生變化�����,具有不同的涂層��、不同的藥物配方�����,二者產(chǎn)品技術(shù)特征不同�����,但參比器械審查時(shí)已考慮了藥物釋放、安全性和有效性��、感染率、生物相容性等問題��,因此該技術(shù)特征差異未產(chǎn)生需考慮的安全有效性新問題��,且有方法可用于評估該技術(shù)特征是否對產(chǎn)品的安全有效性產(chǎn)生影響���。FDA通過審評(例如臺(tái)架�����、動(dòng)物和/或臨床試驗(yàn))來確定差異是否給新組合產(chǎn)品帶來重大的安全性或有效性問題���。這些內(nèi)容對于證明新組合產(chǎn)品與產(chǎn)品A的實(shí)質(zhì)等同和/或符合適用的特殊控制都是必要的,如果未能證明二者的實(shí)質(zhì)等同性���,或者不符合適用的特殊控制��,則二者不具有實(shí)質(zhì)等同性。在這種情況下���,允許510(k)途徑實(shí)質(zhì)等同的方式應(yīng)用于組合產(chǎn)品中藥品部分的單獨(dú)申請���,證明實(shí)質(zhì)等同性和遵守特殊控制足以證明組合產(chǎn)品藥品部分涂層方法變化的安全性和有效性���。
2.4 典型情況4
典型情況4為組合產(chǎn)品藥品部分的濃度降低。假設(shè)參比產(chǎn)品為已上市的產(chǎn)品A��;新組合產(chǎn)品與產(chǎn)品A的藥品部分相同,唯一變化是浸漬到組合產(chǎn)品表面的藥品部分具有較低的濃度(如從500 μg/cm2降低至400 μg/cm2)�����,降低組合產(chǎn)品藥品部分濃度的目的是保持組合產(chǎn)品的有效性的同時(shí)�����,通過減少組合產(chǎn)品的藥物釋放量,降低藥物所致不良反應(yīng)的可能性,但組合產(chǎn)品的預(yù)期用途或標(biāo)簽不變;此外�����,申請人已向FDA提供引用組合產(chǎn)品藥品部分的NDA授權(quán)的文件���。
研討班丨2023藥品委托生產(chǎn)管理與供應(yīng)商審計(jì)策略
對于這種典型情況,F(xiàn)DA認(rèn)為該組合產(chǎn)品的材料和其他特征與參比產(chǎn)品相比發(fā)生了變化,該組合產(chǎn)品具有較低的藥物濃度,與參比產(chǎn)品的技術(shù)特征不同���,但參比產(chǎn)品審查時(shí)已考慮了藥物濃度及感染率相關(guān)的安全有效性問題��,因此該技術(shù)特征差異未產(chǎn)生需評價(jià)的安全有效性新問題��,且有相應(yīng)的方法可評估該技術(shù)特征變化是否對產(chǎn)品的安全有效性產(chǎn)生影響�����。FDA通過審評(必要時(shí)包括臨床數(shù)據(jù))來確定差異性是否給新組合產(chǎn)品帶來重大的安全性或有效性問題��,允許發(fā)生這種改變的組合產(chǎn)品采用510(k)途徑進(jìn)行申報(bào)���。
2.5 典型情況5
典型情況5為組合產(chǎn)品的藥品部分發(fā)生改變(用其他抗菌劑替代其組合產(chǎn)品原有的藥品部分)��。假設(shè)參比產(chǎn)品為已上市的產(chǎn)品A��;申請人將產(chǎn)品A中的抗菌劑A替換為NDA已獲批準(zhǔn)的抗菌劑B(適用于治療急性細(xì)菌性皮膚病和皮膚感染)���,組合產(chǎn)品的適應(yīng)證或使用說明不變;申請人已向FDA提供引用NDA授權(quán)的抗菌劑B的文件���。
對于這種典型情況���,F(xiàn)DA認(rèn)為二者活性成分不同,且新組合產(chǎn)品與參比產(chǎn)品在設(shè)計(jì)��、材料等特征方面也存在很大差異��,引起了組合產(chǎn)品安全有效性的新問題��,新組合產(chǎn)品與參比產(chǎn)品不具有實(shí)質(zhì)等同性。在這種情況下�����,F(xiàn)DA不允許組合產(chǎn)品中藥品部分的單獨(dú)申請采用510(k)實(shí)質(zhì)等同途徑的方式���。組合產(chǎn)品與參比產(chǎn)品的比較���,不能充分證明藥品部分在其新使用條件下的安全有效性(包括新的藥品適應(yīng)證以及藥品與器械的組合使用)。該組合產(chǎn)品在上市之前需要按PMA途徑申請或者可能適合采用De Novo途徑申請��。
3�����、 借鑒意義分析
3.1 中美含藥醫(yī)療器械上市前申報(bào)路徑異同
在FDA��,含藥醫(yī)療器械上市前申報(bào)路徑分為PMA�����、De Novo�����、510(k)3種���,必要時(shí)根據(jù)《組合產(chǎn)品上市前路徑指導(dǎo)原則》第Ⅲ部分的要求��,F(xiàn)DA對于產(chǎn)品申報(bào)路徑選擇原則��,主要是基于產(chǎn)品的風(fēng)險(xiǎn)程度��、新型程度的不同以及組合產(chǎn)品所含藥物成分是否已獲得NDA相應(yīng)適應(yīng)證的批準(zhǔn)等因素進(jìn)行考量�����。
在我國�����,《關(guān)于藥械組合產(chǎn)品注冊有關(guān)事宜的通告》[2](2021年第52號(hào))第二條規(guī)定���,以醫(yī)療器械作用為主的藥械組合產(chǎn)品,應(yīng)當(dāng)按照醫(yī)療器械有關(guān)要求申報(bào)注冊��;同時(shí)���,《醫(yī)療器械分類規(guī)則》[3](國家食品藥品監(jiān)督管理總局令第15號(hào))第六條(四)規(guī)定���,以醫(yī)療器械作用為主的藥械組合產(chǎn)品���,按照第三類醫(yī)療器械管理。根據(jù)上述規(guī)定��,明確了我國的含藥醫(yī)療器械是按照第三類醫(yī)療器械注冊申報(bào)�����,由國家藥品監(jiān)督管理局(以下簡稱為“國家藥監(jiān)局”)醫(yī)療器械技術(shù)審評中心負(fù)責(zé)具體的產(chǎn)品技術(shù)審評工作�����,需要聯(lián)合審評的項(xiàng)目���,注冊申報(bào)資料轉(zhuǎn)國家藥監(jiān)局藥品審評中心進(jìn)行同步審評�����。
3.2 中美含藥醫(yī)療器械注冊申報(bào)要求
在我國�����,對于含藥醫(yī)療器械的注冊申報(bào)基本要求,除應(yīng)按照《醫(yī)療器械注冊申報(bào)資料要求及說明》等相關(guān)要求準(zhǔn)備產(chǎn)品注冊申報(bào)資料外,建議參考國家藥監(jiān)局發(fā)布的通用型指導(dǎo)原則《以醫(yī)療器械作用為主的藥械組合產(chǎn)品注冊審查指導(dǎo)原則》(2022年第3號(hào))[4]等相關(guān)文件的要求���,例如在注冊申請表中注明“藥械組合產(chǎn)品”��,在CH3.8(其他資料)提交藥物相關(guān)產(chǎn)品描述���、藥物和/或醫(yī)療器械與藥物相互作用、藥物含量/劑量選擇資料���,以及相關(guān)研究總結(jié)���,在CH4臨床評價(jià)資料部分根據(jù)產(chǎn)品臨床療效和潛在風(fēng)險(xiǎn)開展臨床評價(jià)等,詳細(xì)內(nèi)容見該指導(dǎo)原則���。該指導(dǎo)原則系對以醫(yī)療器械作用為主的藥械組合產(chǎn)品注冊審查的一般指導(dǎo)文件��,闡述了對該類產(chǎn)品申報(bào)資料的基本要求���,但由于該類產(chǎn)品品種多樣,申請人還需依據(jù)具體產(chǎn)品的特性對申報(bào)資料進(jìn)行充實(shí)和細(xì)化���,具體的產(chǎn)品審評要求分布于各類產(chǎn)品的專用型指導(dǎo)原則中�����,如《冠狀動(dòng)脈藥物洗脫支架臨床前研究指導(dǎo)原則》《冠狀動(dòng)脈藥物洗脫支架臨床試驗(yàn)指導(dǎo)原則》《人工關(guān)節(jié)置換術(shù)用丙烯酸樹脂骨水泥注冊技術(shù)審查指導(dǎo)原則》等���。
含藥醫(yī)療器械各種類型的產(chǎn)品復(fù)雜多樣���,產(chǎn)品自身的風(fēng)險(xiǎn)存在差異,例如冠狀動(dòng)脈藥物洗脫支架和含藥腹腔引流管的風(fēng)險(xiǎn)存在差異��;同類型產(chǎn)品的注冊申報(bào)或變更注冊的不同情形亦存在較大差異���,例如組合產(chǎn)品與同類已上市產(chǎn)品相比藥物種類��、涂層等可能存在差異�����,或變更產(chǎn)品與自身原上市產(chǎn)品相比藥物的含量等可能存在差異��。不同類型的產(chǎn)品風(fēng)險(xiǎn)不同�����,不同變化情形的差異程度和導(dǎo)致的風(fēng)險(xiǎn)亦不同��,因此其對應(yīng)的評價(jià)思路和審評關(guān)注點(diǎn)必然存在差異���。
針對各個(gè)產(chǎn)品不同的具體情況,如何選擇合適的評價(jià)路徑以及如何提交充分的支持資料��,是目前該類產(chǎn)品申報(bào)的難點(diǎn)問題��。FDA的《組合產(chǎn)品上市前路徑指導(dǎo)原則》歸納總結(jié)了含藥醫(yī)療器械5種最典型情況���,分別介紹了不同的變化情形如何選擇合適的申報(bào)路徑及如何選擇參比器械��,為相關(guān)企業(yè)研發(fā)和準(zhǔn)備產(chǎn)品注冊申報(bào)資料提供了較好的細(xì)化分解思路和借鑒方案�����。同時(shí)�����,該指導(dǎo)原則中分析了不同情形下�����,需要考慮差異或變化內(nèi)容給產(chǎn)品安全有效性可能帶來的新增風(fēng)險(xiǎn)��,以及需提交哪些支持資料證明產(chǎn)品的安全有效性�����。
但該指導(dǎo)原則亦存在一定的局限性��,指導(dǎo)原則目前給出的5種典型情況舉例說明��,主要是基于該含藥醫(yī)療器械符合510(k)或De Novo申報(bào)途徑時(shí)�����,對于上市前申報(bào)路徑��、參比器械選擇�����、風(fēng)險(xiǎn)識(shí)別及風(fēng)險(xiǎn)控制措施等方面審評關(guān)注點(diǎn)的建議���,而對于判定需歸屬Ⅲ類PMA管理的含藥醫(yī)療器械或需按指導(dǎo)原則第三部分要求申報(bào)的組合產(chǎn)品���,該指導(dǎo)原則并未對相關(guān)的審評關(guān)注點(diǎn)進(jìn)行詳細(xì)闡述;此外���,舉例中提及的藥品部分都是已獲得NDA批準(zhǔn)的藥物成分�����,且申請人可取得引用相應(yīng)藥品部分NDA文件的授權(quán)���,而對于全新的藥品成分或不能取得授權(quán)等情況,審評的關(guān)注點(diǎn)也未進(jìn)行具體的闡述�����。
3.3 建議
建議我國相關(guān)審評部門可參考《組合產(chǎn)品上市前路徑指導(dǎo)原則》�����,選取典型情況進(jìn)行舉例說明的闡述方式��,結(jié)合各種類型和變化情形的組合產(chǎn)品審評實(shí)例,進(jìn)一步根據(jù)含藥醫(yī)療器械的風(fēng)險(xiǎn)情況��、變化情況等��,梳理歸納總結(jié)更多的具體產(chǎn)品��、具體變化情形的審評要求,特別是梳理不同情形下對于產(chǎn)品安全有效性支持資料的審評關(guān)注點(diǎn)��,形成相應(yīng)的審評框架���、審評要點(diǎn)或共性問題答疑���,并及時(shí)公開發(fā)布,用于指導(dǎo)相關(guān)企業(yè)的組合產(chǎn)品申報(bào)注冊工作��。同時(shí)�����,藥械組合產(chǎn)品涉及多學(xué)科交叉���,產(chǎn)品類型復(fù)雜多樣���,現(xiàn)有的藥品或醫(yī)療器械的樣品制備方法、檢驗(yàn)方法學(xué)、劑量篩選���、安全閾值等研究方法和評價(jià)方式可能不一定完全適用于創(chuàng)新型藥械組合產(chǎn)品�����,需進(jìn)一步完善藥械組合產(chǎn)品方法學(xué)研究��,以逐步形成藥械組合產(chǎn)品方法學(xué)研究和評價(jià)框架體系,為該類產(chǎn)品的安全有效評價(jià)提供技術(shù)支持���。
4、 展望
藥械組合產(chǎn)品由于其獨(dú)特的效果���,具有多元的臨床應(yīng)用前景���,創(chuàng)新產(chǎn)品不斷涌現(xiàn),但在藥械組合產(chǎn)品的研發(fā)�����、生產(chǎn)�����、非臨床研究、臨床研究�����、上市臨床路徑選擇��、上市后監(jiān)管等諸多環(huán)節(jié)需考慮的影響因素復(fù)雜多樣��,使其監(jiān)管和注冊申報(bào)更具挑戰(zhàn)性��,監(jiān)管部門對于藥械組合產(chǎn)品的監(jiān)管理念和技術(shù)審評要求將不斷完善。