摘 要 / Abstract
目的:依據風險管理理論和相關法規(guī)要求��,探索行之有效的醫(yī)療器械注冊自檢風險管理模式,以保證其注冊產品的安全有效�����。方法:以注冊自檢全過程風險管理為研究對象�����,應用風險要素分析和評定�����,找出注冊自檢主要風險環(huán)節(jié)���,進行分析�����、評價�����。結果:不同地域��、不同崗位人員對注冊自檢風險管理認識差異較大���,其差異性遵循了各地產業(yè)發(fā)展特點,也導致在技術審評和現場核查中尺度把握的不同���。結論:醫(yī)療器械注冊自檢風險管理屬于全面質量管理的范疇���,應當從監(jiān)管、企業(yè)���、檢驗檢測層面多方位將其作為完整的管理體系進行全面策劃���、實施與風險管理,才能發(fā)揮預期作用��。
Objective: This study aims to explore an effective risk management model for self-testing in medical device registration based on risk management theory and relevant regulatory requirements. The goal is to ensure the safety and effectiveness of registered products. Methods: The research focuses on the risk management of the whole self-testing process in medical device registration. It employs risk element analysis and evaluation to identify and analyze the key risk points in self-testing. Results: Variations in the understanding of risk management in self-testing for medical device registration exist among individuals from different regions and with different job roles. These differences are influenced by the unique characteristics of industrial development in different places, leading to variations in the scope of technical evaluations and on-site verifications. Conclusion: The risk management of self-testing in medical device registration belongs to the scope of comprehensive quality management. To achieve the desired outcomes, it should be comprehensively planned, implemented, and integrated into a complete management system from regulatory, corporate, and inspection and testing perspectives.
關 鍵 詞 / Key words
醫(yī)療器械�����;注冊自檢���;風險管理�����;應用研究
medical device; registration self-testing; risk control; applied research
醫(yī)療器械注冊產品質量保證與其風險管理有著密切的關系�����。法規(guī)要求包含風險管理內容���,風險管理理論應用于法規(guī)實踐與執(zhí)行的方方面面�����,融入現代質量管理學理論體系框架內理解�����,二者為互為表里��、相輔相成���、互相促進并螺旋推動的關系。2021 年10 月22 日��,國家藥監(jiān)局發(fā)布《醫(yī)療器械注冊自檢管理規(guī)定》[1]���,作為《醫(yī)療器械注冊與備案管理辦法》《體外診斷試劑注冊與備案管理辦法》配套文件�����,允許注冊申請人開展注冊自檢�����,提交的注冊產品檢驗報告可以是申請人的自檢報告�����,也可以是有資質的醫(yī)療器械檢驗機構出具的檢驗報告�����。該規(guī)定旨在進一步加快醫(yī)療器械的上市進程���,繼而更好地釋放醫(yī)療器械產業(yè)創(chuàng)新發(fā)展活力,同時強化注冊申請人的主體責任�����。這也意味著我國醫(yī)療器械監(jiān)管模式逐步與國際慣例接軌��,以促進我國醫(yī)療器械產業(yè)高質量發(fā)展�����。在我國深化審評審批制度改革、鼓勵藥品醫(yī)療器械創(chuàng)新發(fā)展���、深化國務院機構改革和職能轉變的大背景下�����,本研究圍繞新修訂的法規(guī)要求���,結合目前我國醫(yī)療器械法規(guī)體系建設現狀和醫(yī)療器械產業(yè)發(fā)展需求[2],以風險管理為視角分別從完善配套法規(guī)體系���、落實監(jiān)管責任��、落實企業(yè)主體責任3 個方向深入發(fā)掘�����、分析注冊自檢制度的實施在加快醫(yī)療器械上市進程��,釋放醫(yī)療器械產業(yè)創(chuàng)新發(fā)展活力��,強化注冊申請人主體責任要求的實際應用過程中的風險要素���,特別是針對過渡期無相應技術審評依據和現場核查標準�����,以及各地監(jiān)管人員和企業(yè)認識不足的現狀所帶來的風險提出風險管理模型��,并進行定量和定性分析,依據分析評價結果�����,總結風險存在的規(guī)律并深入闡述注冊自檢風險識別�����、分析和控制�����、評價��。
1�����、研究背景、目的及意義
1.1 背景
注冊制度改革為企業(yè)帶來的巨大利好是不言而喻的��,同時基于風險出現的普遍規(guī)律�����,應充分認識到政策改革過渡期新增相應的風險也是必然的�����。這種風險是全方位的�����,以企業(yè)為主體�����,監(jiān)管部門政策把握���、審評尺度衡量的差異都是風險的重要來源��。作為監(jiān)管部門��、技術審評人員�����、企業(yè)三方在共同推動政策改革前行的同時及時做出風險預判并總結風險存在的規(guī)律�����,進行有效地識別��、分析和控制�����、評價有著巨大的現實指導意義���。
本課題組進行了大量的文獻以及資料的查閱收集,并進行了大量的面向醫(yī)療器械生產企業(yè)���、醫(yī)療器械檢驗檢測機構���、醫(yī)療器械技術審評部門的問卷調查和現場調研。對所有的調查問卷進行了統(tǒng)計和分析�����,應用了醫(yī)療器械風險管理基本理論,制定了以醫(yī)療器械注冊自檢風險定性分析和定量分析為主的研究路徑�����,同時以甘肅省醫(yī)療器械生產企業(yè)近年來現場檢查缺陷項分布情況的量化分析為輔助��,采用數據分析法�����、風險管理工具應用�����、專家論證和建立企業(yè)實踐基地的多種結合�����,對注冊自檢風險識別���、分析���、評價��、控制以及剩余風險評價進行了全過程研究��,從而為相關人員對醫(yī)療器械注冊自檢進行有效的風險管理提供一定的參考��。
1.2 目的
依據風險管理基本理論�����,結合法規(guī)要求���,以注冊自檢全過程風險管理為研究對象,應用全過程風險要素分析和評定���,找出注冊自檢主要風險環(huán)節(jié)��,并探索行之有效的風險管理模式,以保證其注冊產品的安全有效��。
1.3 意義
隨著我國醫(yī)療器械創(chuàng)新發(fā)展進入新的歷史時期��,對監(jiān)管科學創(chuàng)新提出更高要求��,特別是在醫(yī)療器械法規(guī)尚不完善的情況下��,監(jiān)管科學與風險管理理論實踐的結合將有助于識別內部和外部環(huán)境因素可能存在的風險隱患,確認風險隱患發(fā)生的概率及危害程度�����,運用統(tǒng)計學��、概率論的方法對風險進行排序���,找出存在的主要風險源和分布規(guī)律�����,為下一步的風險處置提供客觀依據���,從而構建監(jiān)管與企業(yè)雙向落實風險防控體系,以預防�����、承擔�����、規(guī)避��、轉移、轉換���、控制風險��,最大程度地避免�����、降低和預防可能帶來的不利影響��,將風險控制在可接受的范圍內�����。以現有法律法規(guī)���、標準、文件規(guī)定與醫(yī)療器械風險管理相關標準規(guī)定的要求有機融合�����,將風險管理理論應用于注冊(備案)申請人在醫(yī)療器械注冊自檢全過程風險管理���,并對識別出的風險點進行研究分析�����,給出相應的控制要求���。將有助于監(jiān)管人員與申請注冊人(備案人)對相關理論的深化理解,指導企業(yè)建立有效的風險管理模式并提高實際應用風險管理工具的能力��,進一步推動注冊自檢行為的規(guī)范��、有序進行���。
2���、研究方法
2.1 文獻查閱
通過收集相關文件資料,經過分析整理���,建立醫(yī)療器械注冊自檢風險管理基本理論依據�����,確定研究過程���、主要風險來源和風險管理要素��。
2.2 問卷調查
對注冊自檢過程中所有可能涉及的風險進行識別���、梳理,根據風險源���、風險發(fā)生的可能性和風險發(fā)生后的影響程度制定醫(yī)療器械注冊自檢風險要素評定表��,并面向醫(yī)療器械生產企業(yè)�����、監(jiān)管部門�����、檢驗檢測機構��、技術審評部門征求意見�����,對列舉的風險來源進行賦值評分���。利用風險矩陣法進行數據分析,根據分析結果制定具體的管理措施[3]���。
2.3 案例分析
對甘肅省近兩年來依據醫(yī)療器械GMP 及附錄��、現場檢查指導原則開展的生產質量管理體系核查和注冊體系核查主要缺陷分布情況進行統(tǒng)計分析�����,從而對質量管理風險分布范圍界定提供參考�����。
2.4 理論實踐
篩選甘肅省具有代表性的醫(yī)療器械生產企業(yè)作為項目合作單位���,搭建注冊自檢風險管理實踐試點基地。
2.5 專家論證
以現場討論會(也可為線上會議)的形式向醫(yī)療器械監(jiān)管部門��、醫(yī)療器械生產企業(yè)��、行業(yè)協(xié)會專家廣泛征集意見并進行研討得出結論�����。該方法是以遞進式方式分為多個層級進行,以多方位信息輸入的篩選��、分析���、匯總為基礎性方法���,通過實踐驗證和論證結果分析形成最終結果。該方法具有實踐性強�����、可雙向反饋的特點��。
2.6 研究方法小結
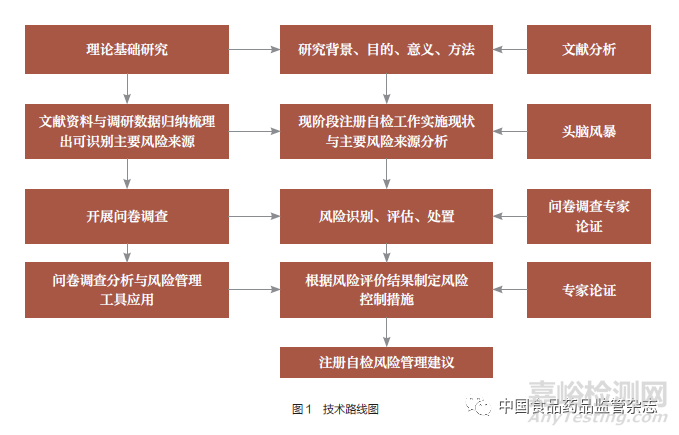
該項目研究內容及技術路線均遵循理論研究與實踐數據驗證相結合的方法�����,主要步驟為:一是通過前期大量的文獻資料收集和相關國內外法規(guī)體系要求的差異分析���,同時同步開展調研活動�����,確定本研究開展的目的�����、范圍�����、方法�����。二是通過研究評價目前國際���、國內檢驗檢測實驗室資質認定評審系統(tǒng)規(guī)范、標準體系��,結合國務院藥品監(jiān)管部門認定(國家級檢驗檢測機構資質認定)中包含但不限于《檢驗檢測機構資質認定條件》�����、《醫(yī)療器械檢驗機構資質認定條件》�����、中國合格評定國家認可委員會認可的規(guī)定及相關強制性國家標準��、行業(yè)推薦性標準體系,以及開展調研意見反饋��,通過頭腦風暴法梳理集中各類風險要素���,確定注冊自檢主要風險來源���。三是依據確定的主要風險來源制定風險評估調查問卷表,設定調查對象及范圍��,并就問卷形式及內容征求行業(yè)內專家意見后隨即開展問卷調查��。四是對收回調查問卷開展數據統(tǒng)計和分析�����,通過定量與定性分析��,得出風險評價結果后制定風險控制措施��,就風險控制有效性組織專家論證會開展剩余風險評價���。通過以上研究路線得出研究結論并提出建議(圖1)��。
3��、研究結果
依據《醫(yī)療器械監(jiān)督管理條例》《醫(yī)療器械注冊與備案管理辦法》《體外診斷試劑注冊與備案管理辦法》《醫(yī)療器械注冊自檢管理規(guī)定》《醫(yī)療器械生產質量管理規(guī)范》法規(guī)要求��,結合YY/T0287—2017 / ISO 13485:2016《醫(yī)療器械 質量管理體系 用于法規(guī)的要求》和YY/T 0316—2016/ISO 14971:2007 更正版《醫(yī)療器械 風險管理對醫(yī)療器械的應用》提供的醫(yī)療器械風險管理基本理論���,同時參考《醫(yī)療器械注冊自檢管理規(guī)定》的具體要求�����, 并結合CNAS-CL01:2018idt ISO/IEC 17025:2017《 檢測和校準實驗室能力認可準則》、RB/T 214—2017《檢驗檢測機構資質認定能力評價 檢驗檢測機構通用要求》��、RB/T 217—2017《檢驗檢測機構資質認定能力評價醫(yī)療器械檢驗機構要求》���,以及2022 年發(fā)布的《醫(yī)療器械注冊質量管理體系核查指南》等規(guī)定��,設置了調查問卷板塊構成�����,從格式上延續(xù)了CNAS-CL01:2018idt ISO/IEC 17025:2017《檢測和校準實驗室能力認可準則》中列出的控制要素分布���。對醫(yī)療器械注冊自檢全過程中風險發(fā)生的概率和風險發(fā)生后對預期結果的影響嚴重程度進行定量的表達和評估,以得到科學的風險定量評價模型�����,用以識別醫(yī)療器械注冊自檢主要風險并研究對應控制措施[4]?�;陲L險源前后相互關系的交互作用模式���,本次調查問卷風險源的征集模型以風險發(fā)生的概率和風險發(fā)生后對預期結果的影響嚴重程度為結構�����,依據本課題組已預估的風險點進行10 分制賦值的方式��,被調查者依據主觀判定逐項進行評分���。基于注冊自檢活動涉及的風險管理模式��、角色�����、資源�����、過程及結果應用評價過程的多維度狀態(tài),調查問卷主要圍繞符合法規(guī)要求��、基于風險管理的醫(yī)療器械質量保障體系為主要視角進行信息采集���。因此��,除監(jiān)管部門��、專業(yè)檢驗檢測機構人員外���,將醫(yī)療器械生產企業(yè)也納為了調查對象,包括企業(yè)研發(fā)人員 ���、質量管理人員、檢驗檢測人員��、生產管理人員��。根據風險矩陣理論基礎��,結合故障樹分析法(FTA)原理并遵循醫(yī)療器械生產質量規(guī)范符合性原則��,明確目的主線后�����,將注冊自檢有效性和符合法規(guī)要求設置為頂事件窗[5],以此推斷注冊自檢實施全過程中人���、機�����、料�����、法��、環(huán)���、測環(huán)節(jié)可能存在的風險并進行邏輯推理,將故障樹中涉及的所有事件以調查問卷形式呈現���,并將問卷中各評價指標進行無量綱化計算�����,即:風險(R)= 風險發(fā)生的概率(P)×風險發(fā)生后的嚴重程度(S)��,得到風險管理研究的綜合指數���、排序和等級��。依據制定的風險管理原則�����,采取不同的風險控制措施并進行有效性評價[6]�����。
調查問卷中共歸納出23 個風險環(huán)節(jié)�����、66 項注冊自檢實施過程中可能存在的風險點,利用風險矩陣法對風險可能發(fā)生的概率及發(fā)生后的嚴重程度進行定量和定性評價[7]���,在全國范圍選擇較有代表性的省市以微信小程序和紙質版形式同時發(fā)放調查問卷��。共收回問卷368 份���,其中有效問卷339 份���,問卷有效率為92.1%。監(jiān)管部門54 份(15.93%)�����, 政府部門下設的醫(yī)療器械檢驗檢測機構72 份(21.24%)��, 醫(yī)療器械生產企業(yè)213 份(62.83%)��。問卷涉及我國廣東���、山東���、上海、重慶�����、山西��、河南�����、海南、云南���、甘肅9 個省份��。
3.1 風險評定結果
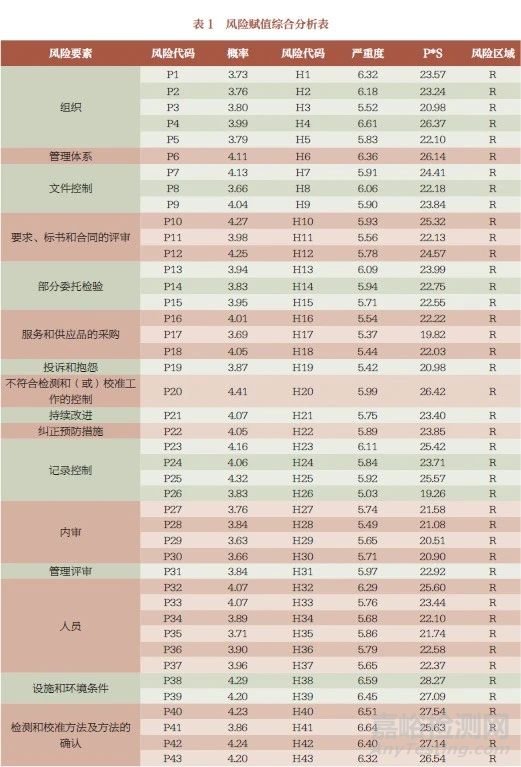
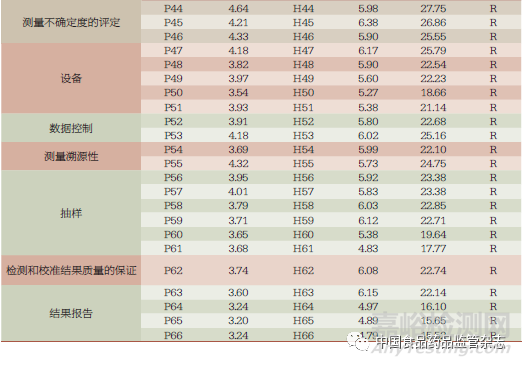
將企業(yè)風險賦值與監(jiān)管風險賦值進行平均���,以保證企業(yè)與監(jiān)管意見的中和。各風險區(qū)域分布情況為:風險范圍10~17 共分布3 項�����、風險范圍17.1~23 共分布32 項���、風險范圍23.1~36 共分布31 項��,均處于中等風險范圍���,需采取相應措施將風險降低至可接受水平,并進行剩余風險評價���。
風險指數排名前十的環(huán)節(jié)依次分布于:設施和環(huán)境條件、測量不確定度的評定、檢測和校準方法及方法的確認���、不符合檢測和(或)校準工作的控制���、組織、管理體系(表1)��。
3.2 風險控制
對以上風險項逐一分別從監(jiān)管角度和企業(yè)發(fā)揮主體責任角度進行原因分析并制定風險控制措施后��,采用邀請行業(yè)內專家評審的方式對剩余風險進行再評價��,確定整體風險是否接受[8]��。針對采取的風險控制措施��,本課題組以行業(yè)內具有代表性的醫(yī)療器械生產企業(yè)作為風險管理措施實踐基地��。本課題組選取部分行業(yè)專家作為風險控制措施有效性再評價方���,對提出的控制措施進行再評價���,同時由甘肅省醫(yī)療器械技術審評部門、檢驗檢測機構���、現場檢查員同步再評價��,整合多方評價要素形成再評價結論�����,用以評價風險管理的有效性���。通過風險控制措施��,各剩余風險發(fā)生概率和風險發(fā)生嚴重程度顯著降低���,說明通過風險管理活動,能夠有效控制重要風險發(fā)生概率�����。
3.3 監(jiān)管人員調查數據分析
對調查問卷進行分組排序與統(tǒng)計分析�����,對收集到的各省��、自治區(qū)�����、直轄市監(jiān)管人員調查數據進行分類統(tǒng)計���,對不同省份��、不同崗位從事醫(yī)療器械檢查審評��、專職檢驗��、監(jiān)管執(zhí)法等人員關于66 項關鍵風險的發(fā)生概率和發(fā)生后的嚴重程度的評估結果進行數據計算分析���,通過橫向對比得到不同省份監(jiān)管人員評估差異,通過縱向對比得到不同崗位監(jiān)管人員評估差異�����。
經統(tǒng)計��,發(fā)現各調查省份66項風險評估值最大值與最小值差異比值平均在1 倍以上��,反映不同省份不同地區(qū)因產業(yè)發(fā)展特點和監(jiān)管理念不同��,對于同一風險要素的發(fā)生概率和發(fā)生后的嚴重程度認知不同���,預估值的差異性較大���。其中��,對風險發(fā)生的概率判斷���,河南與廣東評分值相對較高,重慶與云南估值最低��;對風險發(fā)生后的嚴重程度估值判斷�����,海南與河南估值較高���,山東估值最低��。從事醫(yī)療器械檢查審評���、專職檢驗、監(jiān)督執(zhí)法等不同崗位人員對于注冊自檢風險識別同樣存在差異���,但其差異性遵循了各地產業(yè)發(fā)展特點��,與其崗位關聯程度不高���。以上結果反映出各省��、自治區(qū)、直轄市監(jiān)管人員對于注冊自檢風險識別的差異���,從而導致在技術審評和現場核查中尺度把握的不同�����。產業(yè)較為發(fā)達的省份對特定風險的發(fā)生概率與其產業(yè)發(fā)展是呈正向關聯分布的�����,而對于風險發(fā)生后的嚴重程度認知的不同��,更多的來自監(jiān)管模式與產業(yè)發(fā)展規(guī)模及程度的不同���。本課題組在前期與企業(yè)開展的調研活動中了解到,涉及監(jiān)管層面的顧慮與困惑大部分來自企業(yè)注冊產品在不同省份注冊申請時���,審評人員與現場檢查員在體系核查和資料審查時存在要求不一���、結論不一的情況�����,給企業(yè)實施注冊自檢帶來困擾�����。
3.4 企業(yè)人員調查數據分析
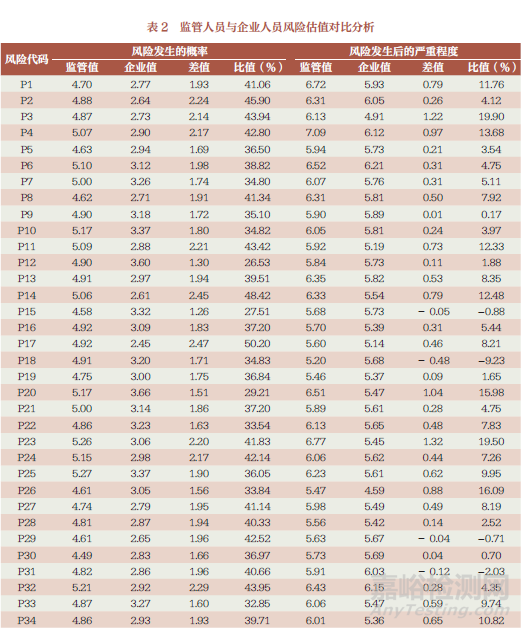
對收集到的來自企業(yè)從事研發(fā)��、生產���、質量管理、檢驗四類崗位管理與工作人員估值信息進行分類統(tǒng)計���,發(fā)現對于同一風險要素發(fā)生概率�����,企業(yè)相關崗位人員估值遠低于監(jiān)管人員��,66 項中有24 項風險比值低于監(jiān)管人員的40% 以上���;風險發(fā)生后的嚴重程度估值與監(jiān)管人員相近(表2)�����。從崗位分布上看���,從事檢驗崗位人員風險估值普遍高于其他崗位人員。
本課題組對監(jiān)管人員和企業(yè)人員風險平均估值進行了逐一對比和分析��,發(fā)現風險發(fā)生的概率存在較大差異的風險環(huán)節(jié)主要分布在:組織機構��、體系文件管理與法規(guī)要求的適用性��、采購評審記錄��、委托檢驗全過程管理�����、采購規(guī)范性�����、記錄的真實完整可追溯性��、內審有效性��、管理評審有效性���、人員配置與能力驗證有效性���、檢驗設備管理、測量溯源性���、抽樣規(guī)范性�����、注冊自檢報告規(guī)范性�����。對風險發(fā)生后的嚴重程度存在的差異比值最大為23.42%���,主要為關鍵人員職責、對不符合項的控制和評價�����、報告中關于型號覆蓋說明的要求。
以上分析可以看出�����,企業(yè)人員主觀意識風險估值要遠低于監(jiān)管人員估值���,由此表明企業(yè)方對注冊自檢風險管理的研究還需進一步加強�����,而作為監(jiān)管人員在充分履職盡責的前提下�����,應當建立綜合評判的能力�����,把握好技術審評工作基本要求和尺度,做到科學界定���、重點明確���、標準統(tǒng)一�����。對于列出的66 項風險因素是廣泛存在于其體系運行過程和日常生產管理及質量控制中���,因此發(fā)生概率較高,對于其產生的后果評估是理性且符合企業(yè)發(fā)展實際的�����。特別是結合當前“放管服”要求�����,以鼓勵藥品醫(yī)療器械創(chuàng)新發(fā)展為背景�����,對風險來源的識別既遵循客觀事實又能理性分析評判其結果影響是非常必要的��。
4���、結 論
本研究以醫(yī)療器械注冊自檢政策實施為研究背景���,應用全面質量管理理念�����,建立了風險管理模型圍繞注冊自檢重要的風險環(huán)節(jié)進行風險管理研究�����,通過定性和定量分析方法進行了基本的風險等級劃分��,并進行了可接受風險的排除���。通過定量評價的結果得出了各風險點所屬風險層級,以及風險分布結論���。根據風險評價結果制定了風險控制措施��,并對擬實施控制措施后預期剩余風險進行了再評價�����。在此過程中對注冊自檢各階段風險評價結果以及剩余風險進行了簡要梳理和說明,通過研究得出如下結論���。
醫(yī)療器械注冊自檢風險管理屬于全面質量管理的范疇��,應當從監(jiān)管�����、企業(yè)、檢驗檢測層面多方位將其作為完整的管理體系進行全面策劃、實施與風險管理���,才能更好地控制可能發(fā)生的風險。從風險評價結果來看,各方對于注冊自檢存在明顯差異��。因此��,本著預防為先的理念�����,各相關方必須建立起全員風險管理意識��,將風險管理要求貫穿于整個產品實現的全過程�����,實施全線管理�����,多方反饋��,以保證產品的風險可控��。企業(yè)作為主體責任人應當進行充分的風險預判���,在不可接受的風險發(fā)生前消除隱患��,以防風險發(fā)生后所帶來的巨大損失���,從而真正實現注冊自檢達到預期目的,增強企業(yè)發(fā)展源動力���。
總體來看���,監(jiān)管人員與企業(yè)人員對于風險發(fā)生的概率與發(fā)生后的嚴重程度認識存在一定的差異。對于風險發(fā)生的嚴重程度評估是理性且符合企業(yè)發(fā)展實際的��,對風險來源的識別是遵循客觀事實且理性的�����。
從法規(guī)層面來看���,我國醫(yī)療器械行業(yè)監(jiān)管法規(guī)體系正在不斷完善中���,配套規(guī)章制度逐步完善,其中的風險是可識別且可控的���;從企業(yè)層面來看��,風險管理是一項需長期堅持的工作���。監(jiān)管層對推出的“政策性公共產品”開展理論和實踐的風險管理研究,并應用于監(jiān)管政策具有重大的意義��。同時��,本次課題研究對于注冊自檢監(jiān)管新工具�����、新方法有效的風險管理具有實踐意義��。
5���、討論及建議
本研究應用全面質量管理理論基礎�����,將醫(yī)療器械注冊自檢政策實施視為公共“產品”的一種��,引入風險管理理念�����,選擇必要的統(tǒng)計學研究方法和風險管理工具��,用于注冊自檢全過程風險識別���、評估與處置��。醫(yī)療器械技術審評機構�����、核查機構��、醫(yī)療器械注冊申請人可以從以下方面完善監(jiān)管法規(guī)體系和注冊自檢質量管理體系�����,以期降低注冊自檢的風險���。
5.1 全面性
醫(yī)療器械技術審評工作是在現行法規(guī)��、標準體系、當前科學認知水平和現有產品技術要求基礎上形成的���,但隨著法規(guī)�����、標準的不斷完善�����,相關科學技術的不斷發(fā)展���,技術審評相關內容及配套文件也需適時進行調整。目前���,與注冊自檢相關聯的審評“技術清單”��、配套法規(guī)���、標準體系均有待進一步完善�����。如何將注冊現場核查與注冊技術審評要求相關聯���,以方法和證據提供的科學性、有效性��、真實性為重點�����,開展全面技術審查��,保證滿足產品技術要求是亟待解決的問題��。
5.2 統(tǒng)一性
醫(yī)療器械技術審查指導原則是對注冊申請人和審查人員的指導性文件�����,適用于企業(yè)準備其醫(yī)療器械產品注冊申報���,同時也適用于審評人員對醫(yī)療器械產品上市前申報資料的審查���?��!夺t(yī)療器械注冊自檢管理規(guī)定》出臺后,由于國家層面尚未出臺相應的技術審查指導原則�����,目前醫(yī)療器械審評人員只能依據審評經驗評估���,因而對技術審評質量的可控性和實效性都形成了一定的制約風險。
5.3 規(guī)范性
為確保不同區(qū)域�����、不同機構審評檢查尺度的統(tǒng)一��,確保醫(yī)療器械注冊自檢政策紅利落到實處�����,一是應盡快研究制定醫(yī)療器械注冊自檢現場檢查指導性文件�����,為檢查員開展現場檢查提供統(tǒng)一的檢查要求,保證不同區(qū)域���、不同能力���、不同機構的檢查員保持較為均一的檢查尺度,以保證技術審評的穩(wěn)定性和可重現性��。二是盡快研究制定醫(yī)療器械注冊自檢技術指南�����,為企業(yè)順利開展自檢工作提供技術指導�����。三是醫(yī)療器械種類繁雜�����、學科門類多樣��、特異性產品多���,應借助專業(yè)技術機構��、醫(yī)療器械行業(yè)協(xié)會���、龍頭企業(yè)��,從具體品種入手���,建立相關產品的注冊自檢規(guī)范。例如��,借助專業(yè)技術機構的力量建立體外診斷試劑注冊自檢規(guī)范��,借助行業(yè)協(xié)會的力量建立軟性親水接觸鏡注冊自檢規(guī)范�����,借助龍頭企業(yè)的力量建立重離子治療系統(tǒng)��、人工心臟瓣膜注冊自檢規(guī)范等���。
5.4 風險管理應用
風險管理是貫穿于醫(yī)療器械全生命周期的活動,其起點是對產品的風險分析���,在分析的基礎上所采用的設計及制造上的技術措施都是為控制風險[9]�����。注冊檢驗屬于醫(yī)療器械從產品設計研發(fā)到推廣應用過程中的一個重要環(huán)節(jié)��,是為了驗證產品臨床評價前的安全性和有效性�����,將產品進行檢驗得出檢驗報告的行為��,因此也應當遵循YY/T 0316—2016/ISO 14971:2007 更正版《醫(yī)療器械 風險管理對醫(yī)療器械的應用》��。醫(yī)療器械生產企業(yè)應當將自檢納入風險管理計劃��,采取有效風險控制措施��。2022 年10 月12 日���,我國發(fā)布醫(yī)療器械風險管理標準GB/T 42062—2022 /ISO 14971:2019《醫(yī)療器械 風險管理對醫(yī)療器械的應用》���。該標準等同采用現行有效的ISO14971:2019 標準, 將于2023年11 月11 日實施��,實施后申請人勢必要調整風險管理策略���,更新風險管理文檔���。監(jiān)管部門應全面分析注冊自檢的開展給技術審評及現場核查工作帶來的全新考驗和風險來源�����,特別是在過渡期應當將該項工作列入風險排查重點工作目錄���,制定符合本地區(qū)監(jiān)管實際的注冊技術審評和現場核查工作要點或指南。
5.5 真實性
此前���,我國醫(yī)療器械產品注冊需提供有資質的醫(yī)療器械檢驗機構出具的全項目檢驗報告���,轉為自檢后,企業(yè)可以制定適用于自身需求的自檢計劃進行內部資源合理分配��,從而大幅縮減檢測周期�����,加快產品上市進程���,自檢政策對企業(yè)研究成果轉化��、加快臨床應用等具有重要意義�����,但是其帶來的風險也不容忽視�����,企業(yè)期望產品快速完成注冊檢驗��,在壓縮檢測周期的同時��,如何保證檢測過程合規(guī)��、真實�����、完整��、可追溯是企業(yè)和監(jiān)管部門需要關注的重點��。
引用本文
楊華,李永紅,王瑞,張鑫衍,方延學.醫(yī)療器械注冊自檢風險管理的應用研究[J].中國食品藥品監(jiān)管,2023(9):56-67.