隨著科技的進步�,新的技術(shù)、設(shè)備�����、新的科技成果越來越多的應(yīng)用在藥品研究生產(chǎn)領(lǐng)域�,對藥品研發(fā)和已上市藥品的質(zhì)量提升起到了重要作用,由此帶來的藥品生產(chǎn)過程中的變更是生產(chǎn)常態(tài)���,也是客觀必然�。生物藥研發(fā)過程中是允許對工藝過程或參數(shù)等進行優(yōu)化的�,這種優(yōu)化不只存在于臨床開發(fā)階段,在藥物上市后也是會經(jīng)常發(fā)生的�。但是對于這種變化所引入的安全性和有效性風(fēng)險需要謹慎評估。無論蛋白藥物還是細胞治療�����,藥學(xué)變更后的可比性研究都不是一項簡單的任務(wù)�����?��?杀刃匝芯康暮诵哪康氖谴_認變更前的非臨床和臨床數(shù)據(jù)適用于變更后的產(chǎn)品���。本文主要就非臨床和臨床橋接研究進行概述(藥學(xué)可比性研究不在本文討論之列)���。
生產(chǎn)工藝改變是不可避免的
通常來講,生產(chǎn)工藝變更越早其實越好�,最好出現(xiàn)的是低風(fēng)險變更。但實際情況是�,工藝變更既有低風(fēng)險也有高風(fēng)險,變更發(fā)生在藥物開發(fā)過程中�,也發(fā)生在藥物上市以后(Process changes happened anytime and anywhere)。分享幾個變更的案例可能更容易理解一些���。
1)重組蛋白:Shingrix(Herpes Zoster Vaccine)
本品主要發(fā)生的藥學(xué)變更內(nèi)容包括:生產(chǎn)規(guī)模擴大�、表達工藝優(yōu)化使抗原表達量提高���、純化工藝優(yōu)化使抗原收率增加、增加pH處理步驟確保病毒充分清除���、增加原液-45℃長期穩(wěn)定性研究�����、生產(chǎn)場地變更�。關(guān)鍵Ⅲ期臨床研究采用工藝變更后樣品開展。
2)抗體:Ilumetri(Tildrakizumab)
本品臨床研究期間���,平行開發(fā)了商業(yè)批原液生產(chǎn)工藝���。工藝一的原液用于非臨床研究。工藝一經(jīng)生產(chǎn)規(guī)模放大后轉(zhuǎn)移到新的設(shè)施生產(chǎn)���。新生產(chǎn)的產(chǎn)品用于臨床Ⅰ�����、Ⅱ期研究���。目前還未涉及到工藝改變,依然使用的工藝一�����。不過�,后續(xù)對工藝一進行了改良,形成了工藝二�。工藝二較工藝一的區(qū)別主要包括:WCB代替了MCB�����、column changes���、制劑處方變更等。關(guān)鍵Ⅲ期臨床研究采用工藝二生產(chǎn)的樣品開展�����。
3)單抗:Bavencia(Avelumab)
這個產(chǎn)品主要經(jīng)歷了兩套生產(chǎn)工藝���。工藝A的樣品用于非臨床和早期臨床研究���。工藝A優(yōu)化后形成工藝B。最初Avelumab的制劑是10 mg/mL���,灌裝8mL�����,用于非臨床研究、臨床Ⅰ/Ⅱ期研究�。為了支持產(chǎn)品商業(yè)化�����,開發(fā)了更高濃度20 mg/mL的制劑�,該制劑通過工藝B生產(chǎn)的原液所得���,并用于后續(xù)所有Ⅲ期臨床研究���,與擬上市制劑是一致的。
以上列舉的是臨床研究階段藥學(xué)工藝的變更���。上市后藥學(xué)變更也是很常見的�。據(jù)統(tǒng)計���,2014年10月以后EMA上市的29個單抗�����,申請了超過400個上市后生產(chǎn)工藝變更���。以藥王Humira為例,自2003年EMA批準上市以來,前后經(jīng)歷了20次生產(chǎn)工藝變更�����,包括擴大生產(chǎn)規(guī)模�����、增加新的生產(chǎn)設(shè)施�、improvement in process controls and robustness等。
藥學(xué)變更隨之出現(xiàn)的問題就是變更前后的可比性研究�。針對這一問題的指南包括ICH Q5E,Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process�。
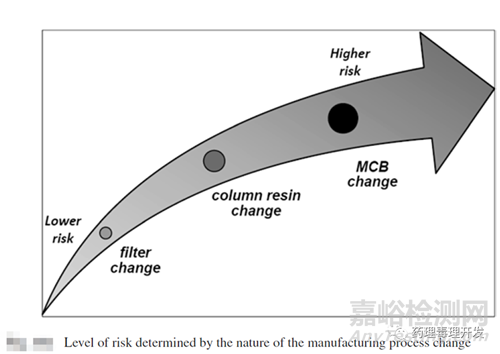
對于工藝變更可比性研究,最先需要做的是判定工藝變更的風(fēng)險等級(Level of risk by type of process change)���。風(fēng)險等級大小直接決定了需要開展的實驗或驗證的數(shù)量���。低風(fēng)險等級的變更,有的可能只需要書面說明合理性���,并不需要開展具體實驗研究���。高風(fēng)險等級可能意味著需要開展大量產(chǎn)品表征、穩(wěn)定性研究,甚至可能的臨床研究���。下圖是個風(fēng)險等級的簡單示意圖。
風(fēng)險等級的劃分有時是有主觀成分在內(nèi)的���。因此���,不同監(jiān)管機構(gòu)EMA、FDA�、WHO對風(fēng)險等級進行了約定。對風(fēng)險的稱謂略有不同�����,但代表的意義類似�。比如對于重大風(fēng)險變更,F(xiàn)DA稱為prior approval supplement���,即需要監(jiān)管機構(gòu)批準后方能執(zhí)行���,EMA則稱為TypeⅡ變更,按數(shù)字進行級別劃分�,WHO與FDA則類似。對于中等風(fēng)險變更,F(xiàn)DA稱為changes being effected in 30days�,即需要等監(jiān)管機構(gòu)30天進行批復(fù),EMA則稱為TypeⅠB變更�����。對于低風(fēng)險等級變更�,F(xiàn)DA稱為annual report,即年度備案�,EMA則稱為TypeⅠA變更。具體哪些改變屬于重大風(fēng)險等級�,內(nèi)容較多,可自行翻閱EMA�、FDA、WHO相關(guān)指南�����。我國CDE依據(jù)風(fēng)險和產(chǎn)生影響的程度由高到低分為:重大變更�����、中等變更���、微小變更三大類�。
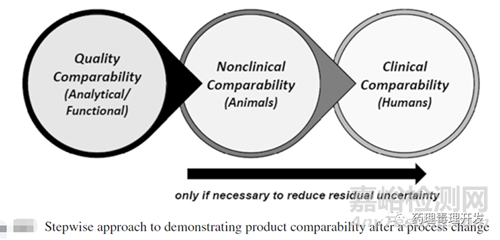
可比性研究通常是分級逐步開展。首先是質(zhì)量對比�����,如果依然存有擔(dān)憂�,則進行非臨床對比,如果還不能解決�����,則進行臨床研究���,如下圖所示。
可比性研究受研發(fā)進程�����、分析方法適用性���、對工藝和藥物的認知程度等影響�����。早期臨床試驗階段���,可比性研究通常不如上市后的廣泛�。因此藥學(xué)變更可分為臨床試驗期間變更和上市后變更兩部分�����。
臨床試驗期間藥學(xué)變更
某些可能對臨床試驗產(chǎn)生重大影響的變更�����,僅藥學(xué)研究可能不足以評估變更帶來的影響���,還需要考慮開展非臨床研究和/或臨床橋接研究�,如新主種子批變更�、特殊輔料變更、延長減毒活疫苗生產(chǎn)用毒種代次�、重新亞克隆篩選等?��!杜R床試驗期間生物制品藥學(xué)研究和變更技術(shù)指導(dǎo)原則》對臨床試驗期間�,對安全性有潛在重大影響的生物制品藥學(xué)變更事項有詳細介紹���,可自行翻閱���。
另外�����,如果可比性研究結(jié)果顯示藥學(xué)變更對臨床試驗的安全性或有效性可能產(chǎn)生負面影響(如改變免疫原性�、產(chǎn)生新雜質(zhì)等)�����,或當(dāng)僅用藥學(xué)分析數(shù)據(jù)無法排除變更可能對有效性和安全性的負面影響時�����,可能需要包括PK/PD和毒理數(shù)據(jù)在內(nèi)的非臨床研究���,甚至需要進行變更前后的臨床橋接研究(參見:臨床試驗期間生物制品藥學(xué)研究和變更技術(shù)指導(dǎo)原則)。
已上市生物制品藥學(xué)變更
《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則》中指出�����,若變更前后產(chǎn)品的生產(chǎn)工藝���、質(zhì)量和穩(wěn)定性研究足以證明可比�����,則無需對變更后產(chǎn)品實施非臨床和/或臨床研究�����。但當(dāng)特定質(zhì)量屬性與安全性和有效性之間的關(guān)系尚未確定�,且觀察到變更前后產(chǎn)品的質(zhì)量屬性存在差異的情況下,應(yīng)實施非臨床和/或臨床橋接性或確證性研究�。非臨床和/或臨床研究的方式和程度應(yīng)結(jié)合藥學(xué)可比性的結(jié)果、對產(chǎn)品性質(zhì)了解的知識水平�、已完成的相關(guān)非臨床和/或臨床研究數(shù)據(jù)以及該藥物的用途,基于具體問題具體分析的原則來確定�����。鼓勵通過藥學(xué)與非臨床的方式開展變更研究�����,若在此基礎(chǔ)上仍無法證明可比性�����,應(yīng)進一步考慮進行臨床研究�����。
某些對生物制品可能產(chǎn)生重大影響的變更,如新主種子批重大變更�����、特殊制劑的關(guān)鍵輔料變更等�,應(yīng)考慮開展非臨床和/或臨床橋接研究。
《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則》第五部分對生物制品常見變更類型及技術(shù)要求有詳細列表說明���,針對哪些變更需要非臨床和/或臨床數(shù)據(jù)支持也有介紹���,內(nèi)容較多,可自行對號入座���,不再贅述。簡單講���,大部分重大變更如表達載體變更�、主種子批變更�����、主細胞庫變更、工作種子批變更���、培養(yǎng)基成分變更���、動物源/非動物源材料變更、變更生產(chǎn)廠房/廠房/生產(chǎn)線�����、替換工藝過程控制參數(shù)和范圍限度�、刪除工藝過程控制參數(shù)和范圍限度、放寬范圍限度���、制劑濃度變更�、制劑體積變更�����、變更制劑處方中輔料組成或濃度���、替換原有輔料���、去除輔料�、佐劑濃度變化等���,應(yīng)考慮進一步開展非臨床和/或臨床的橋接研究���。當(dāng)然,有些變更還要甄別藥學(xué)可比性研究數(shù)據(jù)是否不足以支持變更可比性���。
但其實現(xiàn)有指南還是有些局限性���,只針對哪些變更需要非臨床或臨床數(shù)據(jù)支持,但對于具體需要哪些非臨床試驗或臨床試驗是沒有約定的�����,只是提示根據(jù)藥學(xué)變更前后差異���,決定非臨床或臨床試驗的數(shù)量,是只開展PK研究�,還是PK/PD研究,或是毒理研究�,并未做太多說明。ICHQ5E的2.5部分有簡單介紹非臨床和臨床的考慮,但也比較籠統(tǒng)�����。CDE藥理毒理學(xué)部孫濤老師在2014年一篇文章《橋接研究在藥物非臨床研究與評價中的應(yīng)用》中提到�,針對不同的情況,橋接研究的思路和重點有一定差異�����,但通常是以藥動學(xué)比較研究為先導(dǎo)和主線的���。所以���,PK研究往往是要做的。但由于生物制品藥學(xué)變更復(fù)雜多樣���,CDE鼓勵按照《藥品上市后變更管理辦法(試行)》相關(guān)要求�,通過溝通交流途徑�,就預(yù)期的變更類別、可比性方案和研究內(nèi)容等與監(jiān)管機構(gòu)進行溝通���,特別是對于擬進行重大變更的產(chǎn)品���。
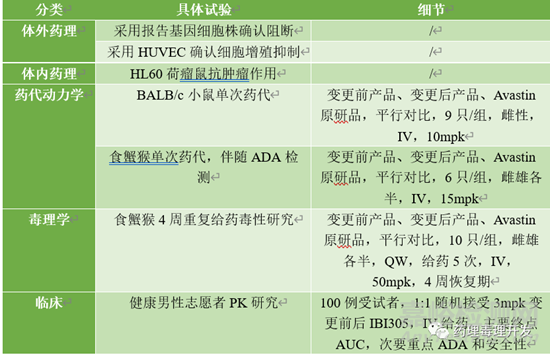
最后分享一個最近信達生物剛發(fā)表的《Comparability strategy and demonstration for post-approval production cell line change of a bevacizumab biosimilar IBI305》�����,屬于已上市生物制品變更細胞株���。當(dāng)然,這肯定屬于重大變更了���。其開展的非臨床和臨床研究如下���。
非臨床橋接的內(nèi)容與藥學(xué)工藝變更的階段、變更的大小密切相關(guān)�����,但藥學(xué)工藝變更通常又是無法避免的�����。先確定變更階段���,是臨床階段還是已上市階段���。再確定變更級別,是重大變更�����、中等變更���、還是微小變更�����,結(jié)合FDA�、ICH�����、EMA���、NMPA的具體指導(dǎo)原則�����,對號入座�����,確定變更對非臨床及臨床研究的影響�,再制定具體的橋接研究計劃。當(dāng)然�����,指南通常只提供大的框架和思路�,詳細研究計劃可以跟監(jiān)管機構(gòu)或業(yè)內(nèi)同行、專家討論�����、溝通�����。