目的:分析國(guó)外客戶對(duì)我國(guó)原料藥生產(chǎn)企業(yè)質(zhì)量審計(jì)過(guò)程中所關(guān)注的重點(diǎn)內(nèi)容,總結(jié)我國(guó)原料藥生產(chǎn)企業(yè)在客戶質(zhì)量審計(jì)中存在的問(wèn)題����,提出解決這些問(wèn)題的方法,為原料藥生產(chǎn)企業(yè)積極應(yīng)對(duì)國(guó)外客戶審計(jì)提供有價(jià)值的建議����。
方法:以我國(guó)某原料藥生產(chǎn)企業(yè)接受國(guó)外客戶GMP審計(jì)為例,分析該企業(yè)在客戶質(zhì)量審計(jì)中存在的問(wèn)題��,對(duì)問(wèn)題剖析后提出相應(yīng)的解決措施和建議��。
結(jié)果與結(jié)論:該原料藥生產(chǎn)企業(yè)在國(guó)外客戶質(zhì)量審計(jì)中涉及的缺陷項(xiàng)目主要分布在質(zhì)量管理�、物料管理、工藝設(shè)備����、廠房與設(shè)施����、文件、生產(chǎn)管理和驗(yàn)證等方面��。企業(yè)應(yīng)當(dāng)加強(qiáng)自檢,積極參與國(guó)外監(jiān)管機(jī)構(gòu)GMP認(rèn)證工作�,從而不斷提高自身質(zhì)量管理水平����。
原料藥(Active Pharmaceutical Ingredients��, API)是生產(chǎn)各類制劑的原料藥物,也是制劑中的有效成分����,其質(zhì)量直接關(guān)系到制劑的質(zhì)量�。長(zhǎng)期以來(lái)�,原料藥一直是我國(guó)醫(yī)藥行業(yè)重要的出口支柱之一��。目前,中國(guó)已成為全球原料藥生產(chǎn)和出口第一大國(guó)����,可生產(chǎn)1500多種化學(xué)原料藥�,產(chǎn)能達(dá)200多萬(wàn)噸��,約占全球產(chǎn)量的1/5以上[1]�。隨著我國(guó)原料藥出口的快速增長(zhǎng)和國(guó)外客戶的增加����,以及藥品市場(chǎng)全球化趨勢(shì)的進(jìn)一步發(fā)展����,越來(lái)越多的國(guó)外客戶開始注重對(duì)其原料藥供應(yīng)商的質(zhì)量審計(jì)����,并將原料藥供應(yīng)商管理納入其企業(yè)生產(chǎn)質(zhì)量管理范疇�。
國(guó)外客戶的質(zhì)量審計(jì)團(tuán)隊(duì)一般由一人至多人組成�,主要來(lái)自其審計(jì)或質(zhì)量部門��,根據(jù)所采購(gòu)原料藥情況��,有時(shí)會(huì)加派其采購(gòu)����、生產(chǎn)����、研發(fā)人員一同參與審計(jì)��。審計(jì)一般為期1~2天��,根據(jù)所采購(gòu)原料藥以及所依據(jù)各國(guó)GMP規(guī)范的不同����,審計(jì)重點(diǎn)也有所不同��,但一般都會(huì)依據(jù)ICH Q7A����,涵蓋GMP六大體系:質(zhì)量管理系統(tǒng)��、設(shè)備設(shè)施��、物料管理�、生產(chǎn)管理、包裝和標(biāo)簽系統(tǒng)以及實(shí)驗(yàn)室管理[2]�,檢查內(nèi)容主要包括供應(yīng)商的廠房、生產(chǎn)設(shè)施�、工藝流程、組織機(jī)構(gòu)����、生產(chǎn)管理人員、文件記錄��、質(zhì)量控制等[3]��,以考查原料藥生產(chǎn)廠家是否具備供應(yīng)符合質(zhì)量標(biāo)準(zhǔn)產(chǎn)品的能力��。
國(guó)外客戶質(zhì)量審計(jì)從所購(gòu)買的產(chǎn)品出發(fā),通過(guò)檢查該產(chǎn)品從物料接收環(huán)節(jié)到成品銷售環(huán)節(jié)來(lái)評(píng)估原料藥生產(chǎn)企業(yè)的質(zhì)量體系����,以此判定其是否為合格供應(yīng)商����。對(duì)于檢查依據(jù),國(guó)外客戶審計(jì)多以ICH Q7A為依據(jù)��,結(jié)合各國(guó)GMP法規(guī)�,對(duì)我國(guó)原料藥生產(chǎn)企業(yè)進(jìn)行質(zhì)量審計(jì)�。
一方面歐美國(guó)家制藥企業(yè)囿于固有觀念對(duì)中國(guó)原料藥生產(chǎn)企業(yè)的生產(chǎn)工藝的受控性和生產(chǎn)記錄的真實(shí)性普遍抱有懷疑態(tài)度��,另一方面國(guó)內(nèi)生產(chǎn)企業(yè)也存在質(zhì)量管理不規(guī)范��、質(zhì)量控制觀念薄弱的問(wèn)題��,質(zhì)量管理依舊是我國(guó)眾多出口企業(yè)較為薄弱的環(huán)節(jié)�,造成國(guó)內(nèi)很多原料藥生產(chǎn)企業(yè)盡管已經(jīng)通過(guò)了FDA��、EDQM等官方機(jī)構(gòu)的現(xiàn)場(chǎng)審計(jì)且注冊(cè)成功,但多數(shù)國(guó)外制劑廠家還是規(guī)定要定期對(duì)原料藥供應(yīng)商進(jìn)行質(zhì)量審計(jì)[4]。因此��,總結(jié)我國(guó)原料藥生產(chǎn)企業(yè)在客戶質(zhì)量審計(jì)中存在的問(wèn)題�,提出解決這些問(wèn)題的方法��,同時(shí)發(fā)現(xiàn)自身不足�,不斷完善質(zhì)量管理體系����,提高產(chǎn)品質(zhì)量標(biāo)準(zhǔn),對(duì)于我國(guó)原料藥出口企業(yè)的長(zhǎng)遠(yuǎn)發(fā)展具有至關(guān)重要的意義。
1��、國(guó)外客戶審計(jì)的主要內(nèi)容
國(guó)外客戶質(zhì)量審計(jì)一般分為現(xiàn)場(chǎng)審計(jì)和文件審計(jì)兩部分����,為期1~2天����,根據(jù)所采購(gòu)原料藥以及所依據(jù)各國(guó)GMP規(guī)范的不同,審計(jì)重點(diǎn)也有所不同�,但一般都會(huì)依據(jù)ICH Q7A,涵蓋GMP六大體系����。原則上,客戶是按照從起始物料到最終產(chǎn)品的原料藥生產(chǎn)順序進(jìn)行檢查��,但是根據(jù)環(huán)境條件和客戶審計(jì)代表的習(xí)慣及專業(yè)背景不同�,檢查有可能從生產(chǎn)的任何一個(gè)工序開始。檢查的重點(diǎn):機(jī)構(gòu)與人員����、廠房和設(shè)施、工藝設(shè)備、物料管理、生產(chǎn)管理�、文件等[5]��。與對(duì)藥品制劑廠家的GMP審計(jì)不同����,一般歐洲藥監(jiān)機(jī)構(gòu)不會(huì)對(duì)非無(wú)菌原料藥廠家進(jìn)行除有因檢查以外的GMP檢查,其只對(duì)制劑廠商進(jìn)行GMP檢查�,而制劑廠商應(yīng)自行進(jìn)行原料藥供應(yīng)商審計(jì),將原料藥供應(yīng)商管理納入其企業(yè)生產(chǎn)質(zhì)量管理范疇。
1.1現(xiàn)場(chǎng)審計(jì)
1.1.1 物料管理
客戶對(duì)倉(cāng)儲(chǔ)的檢查內(nèi)容主要包括原材料的倉(cāng)儲(chǔ)管理和質(zhì)量控制�,成品��、包材����、標(biāo)簽的倉(cāng)庫(kù)管理�,以確保進(jìn)廠的原材料符合要求的質(zhì)量標(biāo)準(zhǔn)并得到良好的儲(chǔ)存�。產(chǎn)品倉(cāng)儲(chǔ)要求分區(qū)明確(分為待驗(yàn)區(qū)��、合格區(qū)、不合格區(qū))����、標(biāo)識(shí)明確��。對(duì)于原材料堆放及發(fā)放要求符合“先進(jìn)先出”原則����,對(duì)原材料包裝容器上的貼簽(包括取樣簽����、待驗(yàn)簽����、放行簽)尤為重視。除此之外�,倉(cāng)儲(chǔ)環(huán)境控制(包括溫濕度控制及蟲鼠防治)也是客戶關(guān)注較多的部分,國(guó)外客戶審計(jì)關(guān)注的重點(diǎn)是看成品區(qū)溫濕度控制是否有24小時(shí)在線監(jiān)控設(shè)備����,如出現(xiàn)溫濕度超標(biāo)情況,是否有迅速的處理流程�。
1.1.2 生產(chǎn)管理
對(duì)于生產(chǎn)工序執(zhí)行情況的確認(rèn)����,客戶首先通過(guò)車間陪同人員的介紹了解基本生產(chǎn)工序�,查看重點(diǎn)操作崗位上的原始記錄,并與現(xiàn)場(chǎng)生產(chǎn)的實(shí)際運(yùn)行情況核對(duì)��。由于原料藥的生產(chǎn)涉及多步反應(yīng)�,工藝較復(fù)雜,客戶一般重點(diǎn)關(guān)注對(duì)產(chǎn)品質(zhì)量影響較大的最后一步�,即在潔凈區(qū)內(nèi)開展的后期精制、干燥�、混粉�、微分、包裝等步驟����。
1.1.3 廠房、設(shè)備和設(shè)施
一般來(lái)說(shuō)客戶對(duì)生產(chǎn)車間廠房��、設(shè)備�、設(shè)施的先進(jìn)性不作過(guò)多要求,而重點(diǎn)關(guān)注下面幾個(gè)方面:廠房的清潔、通風(fēng)和防蟲防鼠措施��;對(duì)潔凈區(qū)則要求廠房密閉性良好��,室內(nèi)外保持適宜壓差�;對(duì)設(shè)備管理,其中尤其重視設(shè)備(如反應(yīng)罐)狀態(tài)標(biāo)識(shí)�,校驗(yàn)合格證的檢查;設(shè)備的清潔周期�、清潔方法、清潔效果驗(yàn)證����,以防止造成交叉污染。
1.1.4 質(zhì)量控制(QC)實(shí)驗(yàn)室
QC實(shí)驗(yàn)室一般是檢查的重點(diǎn)����。客戶在檢查實(shí)驗(yàn)室的過(guò)程中會(huì)持續(xù)關(guān)注各個(gè)儀器的校驗(yàn)情況�,同時(shí)還會(huì)查看有關(guān)設(shè)備儀器校驗(yàn)SOP和使用記錄、校驗(yàn)記錄及設(shè)備儀器是否貼有在有效期內(nèi)的校驗(yàn)合格證��。對(duì)于化學(xué)藥品和試劑��、溶液管理方面�,客戶注重檢查化學(xué)品的使用管理(有效期����、開始使用日期等)��、配制試劑的標(biāo)簽(注明試劑名稱�、配制日期、有效期�、配制人簽字等)等。對(duì)于穩(wěn)定性試驗(yàn)和批留樣����,客戶要查看穩(wěn)定性試驗(yàn)樣品的包裝和儲(chǔ)存方式是否與市售產(chǎn)品保持一致,穩(wěn)定性試驗(yàn)的溫濕度條件����、測(cè)定方法和原始記錄[5]。
1.2文件審計(jì)
文件系統(tǒng)能反映出一個(gè)企業(yè)質(zhì)量管理體系及對(duì)生產(chǎn)過(guò)程的控制水平��,因此客戶審計(jì)時(shí)對(duì)文件系統(tǒng)的檢查往往較為細(xì)致[2]��。對(duì)文件系統(tǒng)的審計(jì)一般針對(duì)文件的受控性��、全面性��、可追溯性��、時(shí)效性來(lái)進(jìn)行�。文件的檢查一般分為現(xiàn)場(chǎng)文件的檢查和文件系統(tǒng)的檢查。
現(xiàn)場(chǎng)文件的檢查主要針對(duì)現(xiàn)場(chǎng)記錄進(jìn)行檢查����,包括:現(xiàn)場(chǎng)操作記錄、設(shè)備日志等現(xiàn)場(chǎng)記錄是否及時(shí)填寫��;記錄中是否出現(xiàn)偏差�,出現(xiàn)偏差后相關(guān)人員是否按照相關(guān)制度進(jìn)行處理、解決�;記錄的領(lǐng)取是否可控等。現(xiàn)場(chǎng)文件記錄應(yīng)保證是最新版本����,如在現(xiàn)場(chǎng)出現(xiàn)了不同版本或舊版本的文件,說(shuō)明文件的發(fā)放����、收回管理出現(xiàn)了問(wèn)題[1]。
對(duì)于文件系統(tǒng)的檢查����,主要關(guān)注驗(yàn)證資料、SOP及其相關(guān)記錄����、批生產(chǎn)記錄等資料��。驗(yàn)證資料主要包括廠房設(shè)施�、設(shè)備��、工藝條件����、清潔、檢驗(yàn)和水系統(tǒng)等�。其中,工藝驗(yàn)證是審計(jì)過(guò)程中客戶重點(diǎn)檢查的對(duì)象��,工藝驗(yàn)證的關(guān)鍵參數(shù)范圍應(yīng)涵蓋實(shí)際生產(chǎn)中的設(shè)定值����。對(duì)于非專用生產(chǎn)線的產(chǎn)品���,清潔驗(yàn)證也是客戶審計(jì)中的重點(diǎn)�。客戶審計(jì)一般都會(huì)對(duì)批生產(chǎn)記錄進(jìn)行檢查�����,客戶會(huì)隨機(jī)或者按過(guò)去購(gòu)買物料的情況抽取2~3批批生產(chǎn)記錄進(jìn)行檢查�。檢查過(guò)程會(huì)核對(duì)生產(chǎn)記錄是否和操作規(guī)程一致,參數(shù)記錄是否規(guī)范以及記錄是否完整�。并以此為追溯線索,檢查相關(guān)物料檢測(cè)情況�����、設(shè)備使用維護(hù)記錄�����、清潔消毒記錄等�。客戶會(huì)重點(diǎn)檢查每一個(gè)步驟的記錄上是否有操作者和復(fù)查人的簽字確認(rèn)�,認(rèn)真檢查原始記錄填寫是否清晰、正確�。
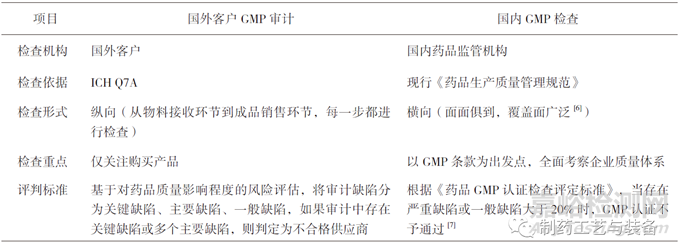
國(guó)外客戶GMP審計(jì)與我國(guó)GMP檢查存在一定的區(qū)別,具體如表1所示���。
表1 國(guó)外客戶質(zhì)量審計(jì)與中國(guó)GMP檢查的區(qū)別
2�、我國(guó)原料藥企業(yè)在國(guó)外客戶審計(jì)中暴露出的問(wèn)題
目前�,國(guó)內(nèi)原料藥生產(chǎn)企業(yè)在接受國(guó)外客戶審計(jì)過(guò)程中普遍暴露出一些問(wèn)題,涉及設(shè)備設(shè)施�����、物料管理、生產(chǎn)管理和質(zhì)量管理體系�。以2013-2014年筆者參與的我國(guó)某原料藥生產(chǎn)企業(yè)24次國(guó)外客戶GMP審計(jì)為例,其審計(jì)報(bào)告中涉及的缺陷項(xiàng)的主要分布情況見(jiàn)表2�����。
表2 我國(guó)某原料藥生產(chǎn)商2013-2014 年客戶審計(jì)缺陷項(xiàng)目出現(xiàn)頻次統(tǒng)計(jì)
結(jié)合該企業(yè)數(shù)據(jù)�、文獻(xiàn)檢索結(jié)果[8-9]以及與行業(yè)內(nèi)其他原料藥生產(chǎn)企業(yè)交流所得資料,在國(guó)外客戶審計(jì)中各個(gè)質(zhì)量管理環(huán)節(jié)涉及到的具體缺陷項(xiàng)目:
1)質(zhì)量管理:檢驗(yàn)用儀器�����、試劑�����、培養(yǎng)基���、標(biāo)準(zhǔn)品�����、對(duì)照品管理不到位���;留樣與穩(wěn)定性研究不全面、不合理���;年度質(zhì)量回顧報(bào)告不全面�、OOS/OOT記錄不全面�。
2)物料管理:物料分發(fā)記錄不完整,倉(cāng)儲(chǔ)庫(kù)房溫濕度記錄未定期整理備份�����。
3)設(shè)備:部分設(shè)備校驗(yàn)后未按規(guī)定發(fā)放校驗(yàn)合格證�����;部分設(shè)備未及時(shí)維修�,或維護(hù)保養(yǎng)不到位;部分生產(chǎn)�、檢驗(yàn)設(shè)備未建立使用、維修���、保養(yǎng)記錄���,記錄不及時(shí)或內(nèi)容不全面���。
4)廠房與設(shè)施:車間、設(shè)備不夠干凈整潔�;生產(chǎn)設(shè)備沒(méi)有定期進(jìn)行維護(hù)保養(yǎng);物料存放區(qū)域面積偏小與生產(chǎn)規(guī)模不匹配���;空調(diào)和水系統(tǒng)未按規(guī)定清潔�����、維修�����、保養(yǎng)�,相關(guān)記錄內(nèi)容不全或記錄不符合文件規(guī)定[10]�。
5)文件:我國(guó)原料藥生產(chǎn)企業(yè)雖然都有建立本企業(yè)生產(chǎn)管理記錄文件系統(tǒng),但大多數(shù)企業(yè)的文件系統(tǒng)并不是很詳細(xì)和規(guī)范[9]���,主要表現(xiàn)在記錄填寫不規(guī)范�、不及時(shí)�;SOP內(nèi)容不完善�;文件之間無(wú)法索引�����;文件歸檔不及時(shí)等�。
6)生產(chǎn)管理:生產(chǎn)現(xiàn)場(chǎng)批記錄未按要求及時(shí)、規(guī)范填寫[9]���;生產(chǎn)現(xiàn)場(chǎng)物料存放不規(guī)范;批生產(chǎn)記錄中未包含部分關(guān)鍵參數(shù)���;部分操作間���、設(shè)備、容器無(wú)狀態(tài)標(biāo)志或標(biāo)志內(nèi)容不全�����,部分操作間�����、設(shè)備狀態(tài)標(biāo)志不能反映生產(chǎn)前后實(shí)際過(guò)程�。
7)驗(yàn)證:中間體檢測(cè)方法未進(jìn)行驗(yàn)證�����;水系統(tǒng)驗(yàn)證未完全實(shí)施���;部分生產(chǎn)設(shè)備、檢驗(yàn)儀器的驗(yàn)證資料不全���,或未以文件形式歸檔保存�;部分驗(yàn)證方案���、驗(yàn)證報(bào)告等驗(yàn)證資料內(nèi)容不完整���。
8)機(jī)構(gòu)與人員:雖然在表2中并未列出,但我國(guó)原料藥生產(chǎn)廠在機(jī)構(gòu)與人員部分面臨的問(wèn)題:培訓(xùn)制度執(zhí)行不到位���,培訓(xùn)計(jì)劃未落實(shí)�;培訓(xùn)內(nèi)容簡(jiǎn)單���、不全面���、針對(duì)性不強(qiáng)�;培訓(xùn)檔案管理不規(guī)范�����,缺少相應(yīng)的培訓(xùn)記錄或記錄收集不完整[8,10-11]�。
9)除以上普遍存在的問(wèn)題外,在近年來(lái)的客戶審計(jì)中還暴露出一些易于被忽略的細(xì)節(jié)問(wèn)題�,如物料標(biāo)簽粘貼不牢;記錄簽字不規(guī)范�;生產(chǎn)設(shè)備標(biāo)識(shí)不全,缺少產(chǎn)品名稱�����、批號(hào)�、數(shù)量等�����。這些細(xì)小問(wèn)題�,常會(huì)在客戶審計(jì)中暴露出來(lái),積少成多���,最終影響審計(jì)結(jié)果的判定�����。
3�、積極應(yīng)對(duì)國(guó)外客戶質(zhì)量審計(jì)的方法
3.1加強(qiáng)自檢
加強(qiáng)自檢,是應(yīng)對(duì)外國(guó)客戶質(zhì)量審計(jì)的重要措施���。以ICH Q7A及各國(guó)GMP規(guī)范為依據(jù)���,從GMP質(zhì)量管理六大體系入手,充實(shí)人員�����,進(jìn)行有針對(duì)性培訓(xùn)���,建立職業(yè)化檢查員隊(duì)伍[8]���,定期組織公司級(jí)、部門級(jí)的自檢���,發(fā)現(xiàn)缺陷立即整改�,并跟蹤后續(xù)整改效果���,提高GMP自檢水平�����,從而不斷完善自身質(zhì)量管理體系�����。在自檢過(guò)程中�,還應(yīng)注重細(xì)節(jié),及時(shí)發(fā)現(xiàn)可能出現(xiàn)的微小缺陷�。對(duì)于非首次審計(jì)的客戶,在接受客戶審計(jì)之前還應(yīng)回顧上次的審計(jì)報(bào)告及對(duì)應(yīng)的糾正和預(yù)防措施(CAPA)報(bào)告�����,確定上次審計(jì)中提到的缺陷項(xiàng)是否已經(jīng)全部整改到位�。
3.2積極參與國(guó)外監(jiān)管機(jī)構(gòu)GMP認(rèn)證工作
由于我國(guó)GMP質(zhì)量體系和ICH Q7A及各國(guó)GMP規(guī)范有一定的區(qū)別�,ICH Q7A 適用于無(wú)菌原料藥在滅菌前的步驟,針對(duì)不同類型品種的生產(chǎn)工藝���,ICH Q7A規(guī)定了需要GMP管理的工藝范圍;而我國(guó)GMP一般適用于藥品制劑生產(chǎn)的全過(guò)程�����、原料藥生產(chǎn)中影響成品質(zhì)量的關(guān)鍵工序�����,在原料藥附錄中著重對(duì)精致�、干燥、包裝工藝的生產(chǎn)環(huán)境提出了要求[12]�。除適用范圍外�,ICH Q7A與我國(guó)GMP在人員、廠房與設(shè)施���、物料、驗(yàn)證�����、文件�����、生產(chǎn)管理���、質(zhì)量管理等方面均有所區(qū)別�����。各原料藥生產(chǎn)企業(yè)通過(guò)進(jìn)行國(guó)外認(rèn)證,了解各國(guó)相關(guān)準(zhǔn)入法規(guī)���,接受FDA�、WHO等國(guó)際組織的檢查���,可以不斷提高自身質(zhì)量管理水平,完善GMP體系�,使企業(yè)自身的質(zhì)量管理水平不斷提高���,達(dá)到提升產(chǎn)品質(zhì)量�、建立品牌良好形象的目的。
4���、結(jié)論
隨著近年來(lái)GMP管理體系的推廣與實(shí)施���,原料藥出口企業(yè)的質(zhì)量體系在不斷的完善,但質(zhì)量管理仍是我國(guó)眾多原料藥出口企業(yè)較為薄弱且不夠重視的環(huán)節(jié)���。加強(qiáng)自檢、積極做好國(guó)外認(rèn)證工作是原料藥生產(chǎn)企業(yè)不斷提高自身質(zhì)量體系水平和更好應(yīng)對(duì)國(guó)外客戶審計(jì)的重要方法���。而在接受國(guó)外客戶質(zhì)量審計(jì)的過(guò)程中�����,我國(guó)原料藥生產(chǎn)企業(yè)可以通過(guò)客戶提出的缺陷項(xiàng)不斷發(fā)現(xiàn)自身的不足,并在提交CAPA報(bào)告時(shí)進(jìn)行整改與完善���,以使自身達(dá)到客戶質(zhì)量要求,從而形成長(zhǎng)期的采購(gòu)關(guān)系�。除此之外,原料藥生產(chǎn)企業(yè)質(zhì)量管理人員在迎檢過(guò)程中�,通過(guò)與客戶的溝通及技術(shù)交流,也是一個(gè)很好的學(xué)習(xí)機(jī)會(huì)�����。積極面對(duì)國(guó)外客戶的質(zhì)量審計(jì)�,對(duì)于我國(guó)原料藥生產(chǎn)企業(yè)不斷完善自身質(zhì)量體系具有至關(guān)重要的意義���。
參考文獻(xiàn)
[1] 溫耀明. 淺談國(guó)外客戶對(duì)原料藥的質(zhì)量審計(jì)[J]. 中國(guó)藥事�,2012�,26(5):534-536.
[2] 趙治華. 原料藥生產(chǎn)企業(yè)對(duì)客戶質(zhì)量審計(jì)的自檢[J]. 中國(guó)藥業(yè)�,2004,13(8):7.
[3] 李玉玲���,楊奕寬. 國(guó)外客戶質(zhì)量審計(jì)對(duì)我國(guó)原料藥企業(yè)出口的影響及對(duì)策研究[J]. 商業(yè)現(xiàn)代化,2008���,(27):18-19.
[4] 梁林美. GMP客戶審計(jì):是難關(guān),也是鍛煉[N]. 醫(yī)藥經(jīng)濟(jì)報(bào)�����,2010-11-08(7).
[5] 孟詠梅�,張淑珍. 淺析FDA對(duì)我公司原料藥的GMP現(xiàn)場(chǎng)檢查[J]. 北方藥學(xué)���,2015�����,12(1):145-146.
[6] 郭睿,秋穎超�,黃晶晶. 各國(guó)GMP現(xiàn)場(chǎng)審計(jì)關(guān)注點(diǎn)差異的比較[J]. 化工管理,2018�����,(10):32-34.
[7] 魏傳波���,顏麗萍,竇學(xué)杰. 中國(guó)���、歐盟對(duì)原料藥GMP檢查方面的比較[J]. 中國(guó)藥事�,2011�,25(2):184-186.
[8] 孫煜�����,呂思伊,魏曼. 湖北省原料藥GMP認(rèn)證中發(fā)現(xiàn)的主要缺陷及建議[J]. 中國(guó)藥事���,2018���,32(9):1264-1270.
[9] 李想,高永寶�,王璐,等. 原料藥企業(yè)藥品GMP認(rèn)證檢查缺陷分析[J]. 品牌與標(biāo)準(zhǔn)化�,2020,(30):62-68.
[10] 洪剛. 原料藥生產(chǎn)企業(yè)GMP質(zhì)量體系缺陷分析與對(duì)策研究[J]. 中國(guó)藥事���,2011�����,25(3):295-300.
[11] 馬東光���,張愛(ài)萍. 2008~2010年我國(guó)生物制品生產(chǎn)企業(yè)GMP檢查缺陷項(xiàng)目分析[J]. 中國(guó)藥事,2011���,25(7):729-739.
[12] 張龍濤���,梁毅. 我國(guó)GMP與ICH Q7A的對(duì)比分析[J]. 西北藥學(xué)雜志�����,2009,24(3):218-220.
本文作者祁洋洋�����,中國(guó)國(guó)際醫(yī)藥衛(wèi)生有限公司�,來(lái)源于中國(guó)藥事,僅供交流學(xué)習(xí)�。