長(zhǎng)效注射混懸劑(微晶或納晶)是將原料藥粉碎或研磨成微米或納米晶體并懸浮于水溶液或油溶液中���,因具有載藥量高和無溶劑殘留風(fēng)險(xiǎn)等優(yōu)勢(shì)���,多用于難溶性藥物的制劑開發(fā),目前長(zhǎng)效注射混懸劑已成為研究熱點(diǎn)之一����。長(zhǎng)效注射混懸劑在慢性疾病治療方面具有優(yōu)勢(shì),近年來在抗精神病藥物中已得到了廣泛應(yīng)用�,已上市產(chǎn)品包括帕利哌酮棕櫚酸酯注射混懸劑、雙羥萘酸奧氮平注射用懸浮液和阿立哌唑長(zhǎng)效注射液均采用微晶或納晶技術(shù)����,因此了解長(zhǎng)效注射混懸劑(微晶或納晶)在體內(nèi)的吸收過程以及吸收影響因素是非常有必要的。
通過查閱相關(guān)文獻(xiàn)得知���,長(zhǎng)效注射混懸劑(微晶或納晶)在機(jī)體內(nèi)的吸收方式主要有兩種���,第一:擴(kuò)散血管吸收(包括溶解和相轉(zhuǎn)移)�����,第二:淋巴系統(tǒng)吸收���;淋巴系統(tǒng)吸收主要包括細(xì)顆粒直接吞噬和局部組織炎癥反應(yīng),藥物通過淋巴系統(tǒng)吸收����,但根據(jù)文獻(xiàn)[1]DARVILLE 等人在研究抗精神病藥物長(zhǎng)效制劑時(shí)通過引流淋巴結(jié)的生物分析中發(fā)現(xiàn)對(duì)于長(zhǎng)效注射混懸劑(微晶或納晶),藥物通過淋巴系統(tǒng)吸收對(duì)于整個(gè)藥物吸收的影響幾乎忽略不計(jì)�����。因此這篇文章我們重點(diǎn)講述長(zhǎng)效注射混懸劑(微晶或納晶)通過藥物擴(kuò)散血管吸收的主要影響因素���。下面我們先講述下肌肉注射和皮下注射的生理結(jié)構(gòu)�����。
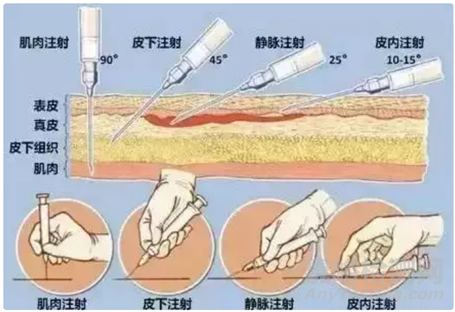
1.皮下注射和肌肉注射的生理結(jié)構(gòu)
皮下注射和肌肉注射的部位一般有豐富的血液和淋巴循環(huán)�����,肌肉注射是將藥物注射到骨骼肌中�,而在骨骼肌中,1mm2的橫切面上�����,有1000以上毛細(xì)血管���,因此肌肉注射給藥的藥物需先經(jīng)注射部位的結(jié)締組織擴(kuò)散,再經(jīng)毛細(xì)血管吸收進(jìn)入血液循環(huán)����。
皮下注射是將藥物注射到皮下組織中,皮下組織有豐富的淺靜脈�����,但其血管少�,血流速度低,因此藥物吸收較慢�。通常情況下肌肉注射比皮下注射吸收要快;通過已上市的長(zhǎng)效注射劑(微晶或納晶)�,我們可以看出,皮下注射或肌肉注射緩釋時(shí)長(zhǎng)可達(dá)一周甚至一個(gè)月及以上���,比如:帕利哌酮棕櫚酸酯注射混懸液����、阿立哌唑注射混懸劑等。下圖1是不同注射給藥途徑的部位���,讓我們更加深入理解不同給藥途徑的位置���。
介紹完皮下注射和肌肉注射的生理特點(diǎn),下面我們來重點(diǎn)講述長(zhǎng)效注射混懸劑(微晶或納晶)吸收影響因素���。
2.影響長(zhǎng)效注射混懸劑(微晶或納晶)吸收因素
2.1原料因素
2.1.1 粒徑
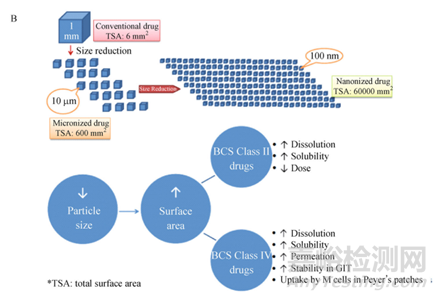
長(zhǎng)效注射混懸劑(微晶或納晶)是將原料藥粉碎或研磨成微米或納米晶體并懸浮于水溶液或油溶液中���,通常情況,粒徑控制是構(gòu)建藥物晶體長(zhǎng)效釋藥系統(tǒng)的基礎(chǔ)���,其理論依據(jù)為經(jīng)典的Noyes-Whitney方程和 Ostwald-Freundlich 方程�,即隨著粒子粒徑的減小���,粒子表面積增大�,藥物溶出速率隨之增加�����,進(jìn)而影響藥物的吸收,下圖2是不同粒徑的總體表面積以及不同粒徑對(duì)藥物溶解度和溶解速率的影響�����。
圖2 Comparison of micro-sized and nano-sized drug crystal in size and distribution in droplets [2]; (B) Advantageous absorption behavior of drug nanocrystals beyond micronized drug
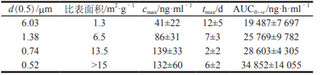
在帕利哌酮的研究資料中得知����,通過研磨法制備了不同粒徑的混懸型注射液��,其比表面積及在 Beagle犬體內(nèi)的藥動(dòng)學(xué)數(shù)據(jù)如表1所示����。由此可見,隨著粒徑的減小�,Cmax和AUC呈明顯增大的趨勢(shì),Tmax呈減小趨勢(shì)�����,但粒徑最小時(shí)的試驗(yàn)結(jié)果偏離了該趨勢(shì)���,可能是因?yàn)榱竭_(dá)到一定程度后����,藥物可能在體內(nèi)聚集,導(dǎo)致接觸面積變小���,進(jìn)而導(dǎo)致粒徑最小時(shí)Tmax結(jié)果出現(xiàn)偏離��。與人體相比�,帕利哌酮在 Beagle 犬體內(nèi)的清除速度較快��,所以顆粒大小對(duì)其在人體內(nèi)藥動(dòng)學(xué)行為的影響比預(yù)期結(jié)果大得多�����。
表1 帕利哌酮的顆粒大小及其肌注后在beagle犬體內(nèi)相應(yīng)的藥動(dòng)學(xué)參數(shù)[3]
2.1.2分子量
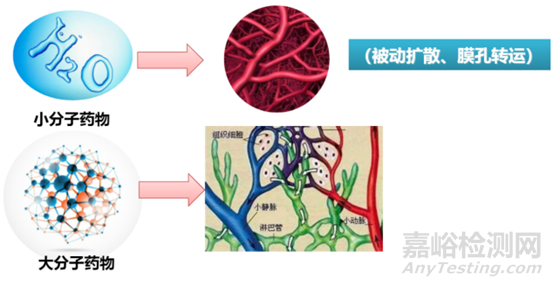
根據(jù)文獻(xiàn)記載�,分子量越大的藥物,吸收也越慢�,大分子(5000~20000)的藥物通過細(xì)胞壁變的十分困難[4],這時(shí)候藥物主要是通過淋巴系統(tǒng)的吸收���,而小分子藥物通常比較容易通過血管壁的微孔�����,這主要是因?yàn)檠鼙谖⒖淄ǔ1容^小�,僅有3nm,因此分子量較小的藥物可以通過�。
2.1.3親脂性和親水性
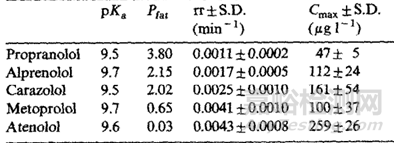
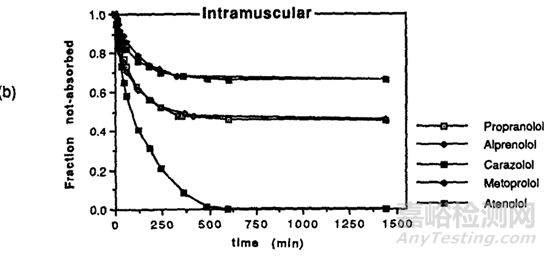
通常情況下,親脂性化合物通過皮下注射和肌肉注射吸收較慢�����,水溶性化合物吸收較快�,這是因?yàn)橹苄运幬锵蚋浇車M織的擴(kuò)散和分配可能會(huì)更慢,當(dāng)然脂溶性藥物有時(shí)也可被淋巴細(xì)胞轉(zhuǎn)運(yùn)�����,但這個(gè)速度也是較慢的�����;根據(jù)文獻(xiàn)[5]介紹���,藥物在體內(nèi)24h的釋放主要取決于藥物的親脂性,親脂性越強(qiáng)的化合物�����,24h的生物利用度越低��,F(xiàn). Kadirr[6]等對(duì)不同親脂性的β受體阻斷劑給豬注射后檢測(cè)剩余藥物的量,其中不同洛爾之間的親脂性見下表2��,結(jié)果顯示(圖3)親水性最強(qiáng)的阿替洛爾吸收最多����,未被吸收的殘余量越少,親脂性最強(qiáng)的普萘洛爾未被吸收的殘余量越多�����。
在早期激素類藥物睪酮的研究報(bào)道中[7]���,在大鼠體內(nèi)�,隨著脂肪酸鏈增加藥物的作用時(shí)間也越長(zhǎng)�����。較長(zhǎng)的酯鏈��,特別是癸酸酯����,會(huì)顯著延遲起效時(shí)間且使血漿 Cmax值變小。短鏈脂肪酸的作用時(shí)間與其分配系數(shù)呈對(duì)數(shù)關(guān)系��,例如應(yīng)用定量構(gòu)效關(guān)系對(duì)南諾龍酯前藥及代謝物的活性進(jìn)行預(yù)測(cè)時(shí),能夠在化合物活性與其在油酸乙酯-水體系中的分配系數(shù)之間建立二項(xiàng)式關(guān)系�����。此外�����,快速測(cè)定藥物在體外的分配系數(shù)有助于預(yù)測(cè)其形成體內(nèi)貯庫(kù)的可能性���。HO[8]等分別用豆蔻酸���、棕櫚酸和硬脂酸合成了恩替卡韋酯類前藥,比較了各產(chǎn)物的水溶性�����、分配數(shù) (logP)���、水解作用和體外釋放曲線,最終選擇棕櫚酸酯前藥為候選藥物�����。
表2 不同β受體阻斷劑理化性質(zhì)及Cmax表[6]
圖3 不同β受體阻斷劑肌肉注射后藥物剩余量
2.1.4晶型
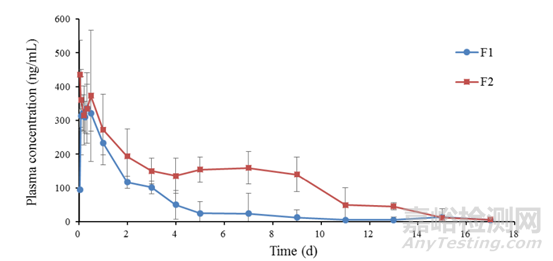
藥物不同的晶型對(duì)藥物吸收也有一定的影響,這可能是因?yàn)椴煌木推淙芙舛炔灰粯?����,?dǎo)致藥物的緩釋作用時(shí)間也會(huì)有一定的影響��。張聰[9]等在美金剛長(zhǎng)效注射劑研究中���,對(duì)晶型I和晶型II進(jìn)行了大鼠肌肉注射��,結(jié)果顯示見下圖4�,晶型I和晶型II兩者緩釋時(shí)長(zhǎng)不一���,其中晶型II的緩釋時(shí)長(zhǎng)明顯長(zhǎng)于晶型I�����。
圖4 經(jīng)肌肉注射給予大鼠125 mg/kg美金剛雙羥萘酸鹽混懸劑(F1(晶型I)和 F(晶型II))的血藥濃度-時(shí)間曲線(mean±SD�����;n = 6)
2.1.5藥物濃度和注射量
通過查詢文獻(xiàn)����,發(fā)現(xiàn)藥物的注射體積和注射量對(duì)藥物吸收無法得到一致的答案,對(duì)于不同的藥物�,不同的載體材料,注射體積或注射量得出的結(jié)論完全不同���,甚至可能會(huì)相反�����。對(duì)于注射水混懸劑�,注射體積越大��,吸收越快���,這主要是因?yàn)樵撍幬镂帐苋芙馑俾视绊?��,即注射體積越大,溶劑被吸收后����,固體藥物越分散����,因此藥物溶解速度快�����,吸收越快���;而對(duì)于親水性藥物水溶液,注射體積越小��,吸收越快��,而對(duì)于不同于油的大分子混懸劑來說����,藥物可能通過溶解以及相轉(zhuǎn)移兩種方式控制,同時(shí)還有一種因素是通過吞噬��,吞噬可能對(duì)于某些細(xì)顆粒直接作用���,但是這種作用幾乎可以忽略不計(jì)���。根據(jù)Leonid Kagan[10]等人對(duì)單克隆抗體的大鼠藥代動(dòng)力學(xué)研究得知,相同給藥量��,大體積皮下注射,藥物初始吸收要快于小體積藥物的吸收�。
圖5 大鼠背部皮下注射藥時(shí)曲線圖(▼— 1 mg/kg、 ∇—10 mg/kg (1 mL/kg)�����、△—10 mg/kg (4 mL/kg)�����、and x— 40 mg/kg)
而HIRANO[11] 等利用動(dòng)物模型建立了一系列藥動(dòng)學(xué)公式��,論證了極難溶藥物的水性混懸型注射液的吸收速率常數(shù) (ka) 與注射體積和濃度的關(guān)系�����,得到注射體積和濃度越大���、ka值越小的結(jié)論����。
2.1.6pH-pKa
據(jù)研究報(bào)道���,pH值在肌肉注射和皮下注射吸收過程中有一定的影響�����,由于組織液的中和或緩沖能力��,有可能會(huì)導(dǎo)致藥物析出����;正常情況下�,肌肉微環(huán)境[20]的pH值為7.4~7.6,為了應(yīng)對(duì)注射導(dǎo)致的創(chuàng)傷��,組織的酸堿度會(huì)在幾個(gè)小時(shí)內(nèi)降至 pH 6.5�,這可能會(huì)影響藥物的溶解度。在所有已知的抗精神病藥LAI制劑中����,藥物分子的 pKa 值為 7~9,在酸堿度下降時(shí)易發(fā)生電離���,而藥物離子化后溶解度普遍會(huì)增加�����,有0.07%的患者在注射奧氮平雙羥萘酸鹽后會(huì)發(fā)生藥物突釋現(xiàn)象���,產(chǎn)生嚴(yán)重不良反應(yīng)[ 13]�����,研究表明���,奧氮平雙羥萘酸鹽在血漿中的溶解度比在pH 7.6緩沖溶液中高15倍,當(dāng)血液滲透到注射部位時(shí)��,藥物會(huì)快速溶解釋放����,導(dǎo)致發(fā)生不良反應(yīng)。
2.1.7 藥物蛋白結(jié)合
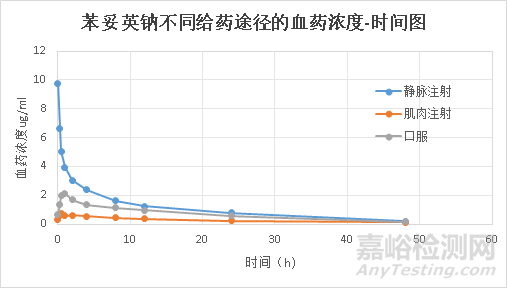
通過文獻(xiàn)得知�����,如果藥物與蛋白結(jié)合很強(qiáng)�����,則藥物在肌肉注射中吸收可能會(huì)慢于口服注射�,比如苯妥英鈉,苯妥英鈉很容易在肌肉內(nèi)與蛋白質(zhì)結(jié)合�,因此該藥物在肌肉吸收要慢于口服����。查詢文獻(xiàn)研究8名正常成午人苯妥英鈉在不同給藥途徑即靜脈注射���、肌肉注射和口服時(shí)的絕對(duì)生物利用度及藥物代謝動(dòng)力學(xué)。肌肉注射的生物利用度比口服低�,峰位僅為口服的三分之一左右(圖6)。
圖6 苯妥英鈉不同給藥途徑的血藥濃度-時(shí)間圖[14]
2.2輔料因素
2.2.1 共溶劑
通常情況���,為了提高藥物的溶解度�,我們會(huì)使用一些共溶劑�,比如乙醇、甘油���、丙二醇和聚乙二醇400�,然而這些共溶劑會(huì)對(duì)吸收起到哪些作用�����,是值得我們研究的���。據(jù)報(bào)道丙二醇���、甘油和聚乙二醇400對(duì)不同化合物的吸收均不同�,有的降低有的提高��。
而乙醇的粘度很小��,在高濃度作用下���,它會(huì)讓注射部位的蛋白變性����,因此它可能會(huì)對(duì)水溶性離子和非離子型藥物如異煙酰胺���、異煙酸甲酯���、異煙酸和鹽酸普魯卡因的吸收有抑制作用,這種影響主要因?yàn)榻Y(jié)締組織通透性的改變�����,最終使其吸收降低��。
據(jù)文獻(xiàn)[15]丙二醇對(duì)鹽酸苯并咪唑吸收的影響說明了共溶劑對(duì)其吸收的影響���,通常情況下���,丙二醇在體內(nèi)吸收慢于水在體內(nèi)的吸收速度���,然而在某些情況下,丙二醇的存在可以防止部分游離堿(或游離酸)的析出���,因此可能會(huì)促進(jìn)藥物的吸收。
2.2.2 表面活性劑
對(duì)于長(zhǎng)效注射混懸劑(微晶或納晶)表面活性劑可以起到潤(rùn)濕劑和穩(wěn)定劑的作用���,目前獲批可用于肌內(nèi)注射的表面活性劑種類并不多��,其中包括吐溫80��、吐溫20��、聚乙二醇4000和泊洛沙姆�����;表面活性劑是注射混懸劑的重要組成部分之一�,它不但可以提高藥物粒徑的穩(wěn)定性�����,還可以增加疏水性藥物被水潤(rùn)濕的能力。據(jù)文獻(xiàn)報(bào)道[16]�����,表面活性劑或親水性聚合物(如聚乙二醇或泊洛沙姆)的吸附會(huì)增加顆粒的親水性并降低內(nèi)化程度�。
2.2.3載體的親脂性
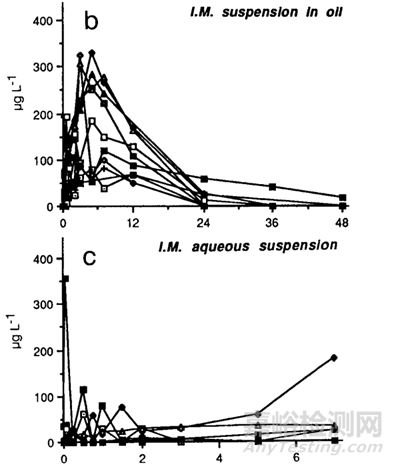
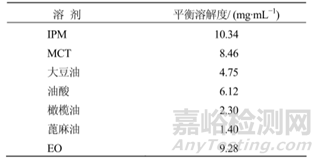
通過文獻(xiàn)[17]發(fā)現(xiàn),將青蒿素制備成油混懸劑和水混懸劑����,分別肌肉注射給10名志愿者,結(jié)果驚奇地發(fā)現(xiàn)����,青蒿素的油混懸劑在體內(nèi)的吸收明顯快于水混懸劑,結(jié)果見下圖7��,其中b為青蒿素油混懸劑���,c為青蒿素水混懸劑:通常情況油混懸或水混懸被認(rèn)為是一種緩釋體系�,而且油在體內(nèi)吸收是相當(dāng)慢的�,有時(shí)候油在體內(nèi)可長(zhǎng)達(dá)一個(gè)月,而本文獻(xiàn)中藥物形成的油混懸劑的吸收快于水混懸劑,主要可能是青蒿素快速溶于油中��,然后再?gòu)挠椭蟹峙涞浇M織液中��,而青蒿素在水中不溶�,因此,沒有油起媒介的作用�����,最終導(dǎo)致青蒿素水混懸劑慢于油混懸劑�����。通過查找文獻(xiàn)[18]得知青蒿素在水中的溶解度小于在油中的溶解度����,青蒿素在水中和不同pH條件下溶解度為60ug/ml~82ug/ml�����,而青蒿素在不同油中的溶解性為1.4mg/ml~10mg/ml�����,在pH7.4的油水分配系數(shù)為1.58。
圖7 青蒿素油混懸劑和水混懸劑肌肉注射的藥時(shí)曲線圖[17]
表3青蒿素在不同油體系中的溶溶解度[18]
2.3臨床給藥位置
2.3.1注射深度
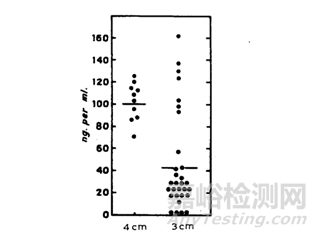
注射不同的深度��,其機(jī)體環(huán)境也有一定的差異���,比如皮下注射脂肪結(jié)締組織和脂肪細(xì)胞較為豐富����,而脂肪組織和肌肉組織的親脂性也是有一定差異��,通常情況���,脂肪組織比肌肉組織的親脂性更強(qiáng)����,血流量也更少�����。據(jù)Dundee[19]等人研究����,將地西泮注射液進(jìn)行肌肉注射,注射深度分別為3cm和4cm��,結(jié)果(結(jié)果見下圖8)顯示,注射3cm的血藥濃度小于注射4cm��,研究人員解釋���,3cm的脂肪層多于4cm��,因此地西泮在3cm吸收更慢���。
圖8 不同注射深度對(duì)地西泮的吸收影響[19]
2.3.2注射部位
注射部位對(duì)藥物的吸收也有一定作用,經(jīng)常使用的部位有臂三角肌�、臀大肌、股四頭肌以及前臂腹側(cè)的肌肉�,據(jù)相關(guān)文獻(xiàn)報(bào)道,與臀肌注射相比�����,三角肌注射時(shí)藥物吸收速度更快����。
3.總結(jié)
綜上所述��,影響長(zhǎng)效注射混懸劑(微晶和納晶)原因非常多���,包括原料自身因素�����、輔料因素以及臨床給藥位置��。原料自身因素包括分子量���、親脂性����、粒徑�����、晶型���、藥物濃度和注射量�����、pH-pKa�����、藥物蛋白結(jié)合等性質(zhì)��,因此在選擇藥物時(shí)需要考慮這些因素��,同時(shí)還需要考慮半衰期和劑量等�,這樣可為立項(xiàng)提供一定的參考;輔料因素包括:共溶劑��、表面活性劑以及輔料的親脂性���,這些均是我們研發(fā)過程中需要考慮的問題���。與此同時(shí),我們可能還需要關(guān)注臨床應(yīng)用���,包括注射位置���、注射速度等。
參考文獻(xiàn):
[1]DARVILLE N, VAN HEERDEN M, ERKENS T, et al.Modeling the time course of the tissue responses to intramuscular long-acting paliperidone palmitate nano-/ microcrystals and polystyrene microspheres in the rat [J]. Toxicol Pathol, 2016, 44(2): 189-210
[2]Tzu-Lan CHANG等 Nanocrystal technology for drug formulation and delivery Front. Chem. Sci. Eng. 2015, 9(1): 1–14
[3]FRANÇOIS M K J, DRIES W M A C, BASSTANIE E D G.Aqueous suspensions of submicron 9-hydroxyrisperidone fatty acid esters: US, 6555544 [P].2003-04-29
[4]魏樹禮《生物藥劑學(xué)和藥物動(dòng)力學(xué)》
[5]J. Zuidema *, F. Kadir, H.A.C. Titulaer, C. Oussoren Release and absorption rates of intramuscularly and subcutaneously injected pharmaceuticals (II) International Journal of Pharmaceutics 105 (1994) 1X9-207
[6]F. Kadirr等人.Drug lipophilicity and release pattern of some /3-blocking agents after intra-adipose injection in pigs International Journal of Pharmaceutics, 64 (1990) 171-180
[7]VAN DER VIES J.Implications of basic pharmacology in the therapy with esters of nandrolone [J].Acta Endocrinol Suppl (Copenh), 1985, 271: 38-44.
[8]HO M J, LEE D R, IM S H, et al.Microsuspension of fatty acid esters of entecavir for parenteral sustained delivery [J].Int J Pharm, 2018, 543(1/2): 52-59
[9]張聰?shù)热嗣澜饎傞L(zhǎng)效注射劑的研究 江南大學(xué) 2021
[10]Leonid Kagan等 Subcutaneous Absorption of Monoclonal Antibodies: Role of Dose, Site of Injection, and Injection Volume on Rituximab Pharmacokinetics in Rats Pharm Res (2012) 29:490–499
[11]HIRANO K, ICHIHASHI T, YAMADA H.Studies on the absorption of practically water-insoluble drugs following injection. III. Intramuscular absorption from aqueous nonionic surfactant solutions in rats [J].Chem Pharm Bull, 1981, 29(3): 834-843
[12]GOMENI R, LAFFONT CM, HEIDBREDER C. A dose switching simulation analyses from Invega® Sustenna® or Risperdal® Consta® to RBP-7000, a new sustained-release formulation of risperidone [J].Clin Pharmacol Ther, 2014, 55(1): 93-103.
[13]MCDONNELL D P, DETKE H C, BERGSTROM R F, et al.Post-injection delirium/sedation syndrome in patients with schizophrenia treated with olanzapine long-acting injection, II: investigations of mechanism [J].BMC Psychiatry, 2010, 10: 45.
[14]霍治平等正常人苯妥英鈉不同給藥途徑 ( 靜脈�、肌肉、口服)的藥物動(dòng)力學(xué)研究
[15]Cheng-Der, Y. and Kent. J.S., Effect of propylene gycol on subcutaneous
absorption of a benzidamole hydrochloride. j. Pharm. SCi.. 71 (1982) 476-478.
[16]BAERT L, VAN 'T KLOOSTER G, DRIES W, et al. Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment [J].Eur J Pharm Biopharm, 2009, 72(3): 502-508.
[17]Titulner, H.&C., Zuidcma, J.. Kager. P.A.. Wetsteyn. J.C.F.M.. Lugt. C.B. and Merkus. F.W.H.M.. The pharmacokinetics of artcmisinin after oral, intramuscular and rectal administration to volunteers. J. Pharm.pharmacol, 42 (1990b)810-813.
[18]高偉祺等蒿素在不同溶劑中平衡溶解度和表觀油水分配系數(shù)的測(cè)定 Drug Evaluation Research 第36卷第1期 2013年02月
[19]J. Zuidema Release and absorption rate aspectsof intramuscularly injected pharmaceuticals International Journal of Pharmaceutics, 47 (1988) 1-12
[20]丁靜雯等影響注射用長(zhǎng)效納米晶混懸型注射液體內(nèi)外釋藥因素的概述中國(guó)醫(yī)藥工業(yè)雜志 Chinese Journal of Pharmaceuticals 2020, 51(12)